| Identification | More | [Name]
Gefitinib | [CAS]
184475-35-2 | [Synonyms]
AKOS 91371
GEFITINIB
IRESSA
n-(3-chloro-4-fluoro-phenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine
N-(3-Chloro-4-fluorophenyl)-7-methoxy-6-[3-(4-morpholinyl)propoxy]-4-quinazolinamine
ZD 1839
4-Quinazolinamine, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-[3-(4-morpholinyl)propoxy]-
Gefitinib(TINIBS)
6-(Benzyloxy)-7-methoxyquinazolin-4(3H)-one
Gefinitib | [EINECS(EC#)]
643-034-7 | [Molecular Formula]
C22H24ClFN4O3 | [MDL Number]
MFCD04307832 | [Molecular Weight]
446.9 | [MOL File]
184475-35-2.mol |
| Chemical Properties | Back Directory | [Appearance]
Light-Yellow Crystalline Powder | [Melting point ]
119-1200C | [Boiling point ]
586.8±50.0 °C(Predicted) | [density ]
1.322±0.06 g/cm3(Predicted) | [storage temp. ]
room temp | [solubility ]
Soluble in DMSO (up to 40 mg/ml) or in Ethanol (up to 4 mg/ml). | [form ]
powder | [pka]
7.00±0.10(Predicted) | [color ]
white to beige | [Usage]
An antineoplastic | [Stability:]
Stable for 2 years from date of purchase as supplied. Solutions in DMSO or ethanol may be stored at -20°C for up to 1 month. | [CAS DataBase Reference]
184475-35-2(CAS DataBase Reference) |
| Hazard Information | Back Directory | [Description]
Gefitinib(184475-35-2) was introduced in Japan as a daily oral monotherapy for the treatment of
inoperable or recurrent non-small cell lung cancers (NSCLC). This anilinoquinazoline
derivative can be synthesized in 6 steps starting from 6,7-dimethoxyquinazolin-4(3H)-one
by successive monodemethylationlacetylation of the 6-hydroxy-group followed by
chlorination and reaction with 3-chloro-4-fluoroaniline, finally deacetylation and alkylation
with 3-(4-morpholinyl)propylbromide complete the synthesis. Gefitinib reversibly inhibits
the activity of the epidermal growth factor receptor tyrosine kinase (EGRF TK). This
inhibits autophosphorylation of EGRF and blocks the cascade of intracellular events which
have been implicated in the proliferation, survival and metastasis of cancer cells. Gefitinib
diplays good selectivity for the EGRF TK relative to other growth factors in human
umbilical endothelial cells. It is similarly selective relative to other kinases, for example cerB2.
Data from two large phase II studies in patients with pretreated NSCLC have shown
that gefitinib induces a response rate approaching 20% in patients receiving the agent as a second line therapy and approximately 10% in those pretreated with more lines of
chemotherapy. Gefitinib has good bioavailability and is metabolized in the liver via the
cytochrome P450 3A4 enzyme system with a mean elimination half life of 28 h. Gefitinib
has been generally well tolerated in cancer patients with predominant side effects being
acne-like skin-rash, diarrhea, nausea, vomiting and mild to moderate myelosuppression.
.
| [Chemical Properties]
Light-Yellow Crystalline Powder | [Originator]
Astra Zeneca (UK) | [Uses]
Gefitinib (Iressa, ZD-1839) is an EGFR inhibitor for Tyr1173, Tyr992, Tyr1173 and Tyr992 in the NR6wtEGFR and NR6W cells with IC50 of 37 nM, 37nM, 26 nM and 57 nM, respectively. | [Uses]
Gefitinib is an antineoplastic. | [Definition]
ChEBI: Gefitinib is a member of the class of quinazolines that is quinazoline which is substituted by a (3-chloro-4-fluorophenyl)nitrilo group, 3-(morpholin-4-yl)propoxy group and a methoxy group at positions 4,6 and 7, respectively. An EGFR kinase inhibitor used for the treatment of non-small cell lung cancer. It has a role as an epidermal growth factor receptor antagonist and an antineoplastic agent. It is an aromatic ether, a member of monochlorobenzenes, a member of monofluorobenzenes, a secondary amino compound, a tertiary amino compound, a member of quinazolines and a member of morpholines. | [Indications]
Iressa (ZD1839) is an orally active tyrosine kinase inhibitor
selective for the epidermal growth factor (EGF)
receptor tyrosine kinase. Iressa is undergoing clinical
trials in the treatment of various solid tumors, including
head and neck cancer, breast cancer and non-small cell
lung cancer. Its antitumor activity is derived from the
fact that the EGF receptor and EGF signaling are
frequently overactivated in sensitive tumors. The major
side effects include diarrhea and skin rash. Bone marrow
toxicity has not been a dose-limiting problem. | [Indications]
The EGFR or ErbB1 inhibitor gefitinib (Iressa(R), AstraZeneca) was originally approved by the US FDA in 2003 under accelerated regulations for the treatment of locally advanced or metastatic non-small cell lung cancer (NSCLC) after progression on docetaxel- and platinum-based chemotherapy. AstraZeneca voluntarily withdrew gefitinib from the market in 2005, owing to failed verification of clinical benefit during post-approval studies. In July 2015, FDA reinstated the approval of gefitinib for a different group of patients (i.e., NSCLC patients with EGFR mutations).
Other approved kinase inhibitors targeting the ErbB family, which includes ErbB1/EGFR, ErbB2/human epidermal growth factor receptor 2 (Her2), ErbB3/ Her3, and ErbB4/Her4, are erlotinib (Tarceva(R), OSI Pharm.), lapatinib (Tykerb(R), GlaxoSmithKline), vandetanib (Caprelsa(R), AstraZeneca), afatinib (Gilotrif(R), Boehringer Ingelheim) , and osimertinib (Tagrisso(R), AstraZeneca). All approved EGFR family inhibitors share a common quinazoline scaffold with the exception of osimertinib, which has a pyrimidinylphenylamine scaffold that resembles that of imatinib and nilotinib. Gefitinib and vandetanib adopt the type I binding mode with “DFG-in” and αC-helix “in” conformation, while erlotinib and lapatinib bind to“DFG-in”with the αC-helix adopting an “out” conformation. Afatinib and osimertinib are covalent inhibitors with an electrophilic enone moiety. | [Brand name]
Iressa (AstraZeneca). | [General Description]
Geftinib(184475-35-2) is available as 250-mg tablets for oral administrationin the treatment of NSCLC for those patients who have failedto respond to platinum-based therapies and docetaxel and hasalso been used against squamous cell cancers of the head andneck. The agent is an inhibitor of the TK of EGF-R and possiblyother TKs as well. Gefitinib is both a substrate and inhibitorof Pgp and BCRP. The agent is absorbed slowly afterbeing administered orally with 60% bioavailability.Metabolism occurs in the liver and is mediated primarily byCYP3A4 to give eight identified metabolites resulting fromdefluorination of the phenyl ring, oxidative-O-demethylation,and multiple products arising as a result of oxidation of themorpholine ring. The O-demethylated product represents thepredominate metabolite and is 14-fold less active comparedwith the parent. The parent and metabolites are eliminated inthe feces with a terminal elimination half-life of 48 hours.The drug appears to be well tolerated with the most commonlyreported side effects being rash and diarrhea. It mayalso cause elevations in blood pressure especially in those patientswith preexisting hypertension, elevation of transaminaselevels, and mild nausea and mucositits.
| [Biological Activity]
Orally active, selective inhibitor of EGFR tyrosine kinase (IC 50 = 23-79 nM). Shows minimal activity against ErbB2, KDR, c-flt, PKC, MEK and ERK-2. Blocks EGFR autophosphorylation and inhibits tumor growth in mice bearing a range of human xenografts. | [Biochem/physiol Actions]
Gefitinib(184475-35-2) is a selective epidermal growth factor receptor tyrosine kinase (EGFR TK) inhibitor. Gefitinib has antineoplastic activity, and has been approved for the treatment on non-small cell lung cancer (NSCLC).Gefitinib has a higher affinity for ATP (adenosine triphosphate) binding site in the EGFR tyrosine kinase domain than ATP. Hence, gefitinib is known to inhibit the progression of endometrial cancer.
| [Clinical Use]
Tyrosine kinase inhibitors:
Treatment of non-small cell lung cancer | [Synthesis]
A mixture of 4,5-dimethoxyanthranilic acid
(133) and formamide was heated to generate the cyclized
quinazoline 134. The quinazoline was selectively monodemethylated with methionine in refluxing methanesulfonic
acid to afford 135 in 47% yield. Compound 135 was
acylated to give acetate 136, which was treated with
refluxing thionyl chloride to yield chloropyrimidine 137.
Chloride 137 was condensed with 3-chloro-4-fluoroaniline
(138) in refluxing IPA to yield anilinoquinazoline 139 in
56% yield from 136. The acetate protecting group in
compound 139 was hydrolyzed with ammonium hydroxide
in methanol, and the free phenol was alkylated with 3-(4-
morpholinyl)propyl chloride (140) to give gefitinib (13) in
55% yield.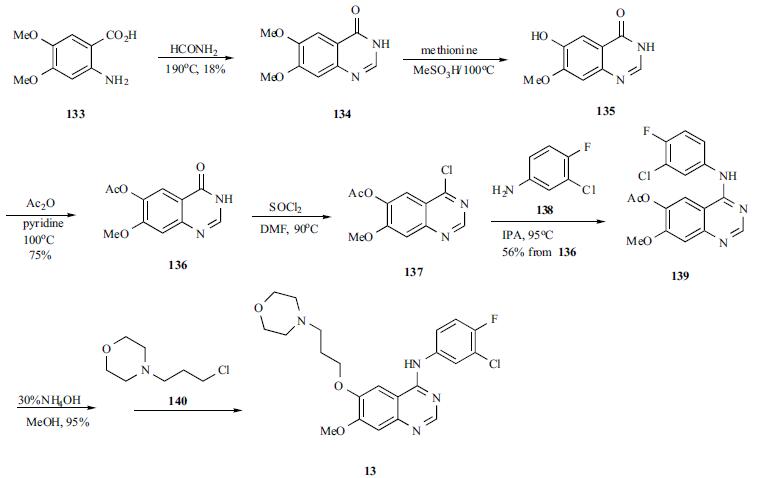
| [Drug interactions]
Potentially hazardous interactions with other drugs
Antibacterials: Avoid with rifampicin (reduced
gefitinib concentration).
Anticoagulants: possibly enhanced anticoagulant
effect with warfarin
Antipsychotics: avoid with clozapine (increased risk
of agranulocytosis).
Antivirals: avoid with boceprevir.
Ulcer-healing drugs: concentration reduced by
ranitidine.
Avoid concomitant use with other inhibitors or
inducers of CYP3A4. Dose alterations may be
required. | [Metabolism]
Extensively metabolised in the liver, mainly by the
cytochrome P450 isoenzymes CYP3A4 and CYP2D6;
the major metabolite is O-desmethylgefitinib, which is
much less potent than gefitinib, and unlikely to contribute
to its clinical activity.
Gefitinib is excreted mainly as metabolites via the faeces
(86%); renal elimination of gefitinib and its metabolites
accounts for <4% of the dose. | [storage]
Store at RT | [References]
1) Baselga et al. (2000), ZD1839 (‘Iressa’) as an anticancer agent; Drugs, 60 33
2) McKillop et al. (2005), Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor; Mol. Cancer Ther., 4 641
3) Sirotnak et al. (2000), Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase; Clin. Cancer Res., 6 4885
4) Ciaradiello et al. (2001), Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor; Clin. Cancer Res., 7 1459 |
| Questions And Answer | Back Directory | [Binding Mode]
The key interactions of gefitinib with EGFR as determined from X-ray crystallographic data are summarized. It binds to the ATP�binding pocket of the active form of EGFR, with the quinazoline N1 in the rear of the ATP-binding pocket, where it hydrogen bonds with the main chain amide of Met793 in the hinge region. Unlike most kinase inhibitors, gefitinib forms only a single hydrogen bond to the hinge. The 3-chloro-4-fluoro aniline substituent extends into the back of the hydrophobic pocket which serves as the ATP-binding cleft. The ortho-fluoro activated chlorine atom attracts the backbone carbonyl oxygen atom of Leu788 within the active site of EGFR and forms a halogen bond. The methoxy group in the 7 position of the quinazoline is in van der Waals contact with Gly796, while the 6-propylmorpholino group extends into solvent. As a type I inhibitor, gefitinib does not occupy the back cleft.
 | [Indications and Uses]
Gefitinib is an antineoplastic target therapy drug with relatively high specificity that was developed by the British pharmaceutical company AstraZeneca; it is the first molecular targeted drug to be used in non-small cell lung cancer treatment. Epidermal growth factors (EGF) are a kind of polypeptide with a relative molecular mass of 6.45x103, and they can bind with epidermal growth factor receptors (EGFR) on target cell membrane surfaces to trigger biological effects. EGFR is a type of tyrosine kinase (TK) type receptor, so when bound with EGF, it will promote TK activation in the receptor. This will cause tyrosine residue in the receptor to autophosphorylate and send continuous dividing signals into the cell, causing cell proliferation and differentiation. EGFR is abundant in human tissue, and it is highly expressed in malign tumors. Gefitinib blocks the signal transduction pathway of cell surface EGFR to prevent tumor growth, metastasis, and growth in blood vessels, and it can induce tumor cell apoptosis. Gefitinib is mainly used to treat non-stem cell lung cancer.
| [Pharmacokinetics]
It is orally effective, with relatively slow absorption and metabolism following intake. The bioavailability of a single oral 250mg dose is nearly 60%, and its area under curve (AUC) is dependent on dosage. With single daily dosages, blood concentration is steady after 7-10 days, with blood concentration peaking 3-7 hours after medication and then gradually following biphasic reduction (its half-life is 12-58 hours, at an average of 28 hours). It is observed as dose-dependent pharmacokinetics, and following multiple dosages, AUC and Cmax increased proportionally. When taken with food, its Cmax and AUC did not decrease significantly. Its plasma protein binding rate is nearly 90%. Gefitinib is metabolized through many different pathways in the livers in a relatively complicated process; the main component of its oxidative metabolism is the cytochrome P450 enzyme CYP3A4, which mainly metabolizes O-Desmethyl metabolites. Metabolites are unrelated to the pharmacological effects of the original drug. The original drug and many metabolites are mostly passed through the biliary tract and excreted through feces, while the amount of drug excreted through urine is less than 4% the original dosage amount.
| [Adverse Reactions]
Gefitinib is relatively well-tolerated, and most negative reactions are mild and reversible, characteristics that are vastly different from those of standard negative reactions to cytotoxic drugs. Common negative reactions include diarrhea, nausea, rashes, acne, vomiting, and feebleness. Only 1% of patients have had to cease treatment due to negative reactions with an occurrence rate over 20%. There have also been rare cases of acute interstitial pneumonia.
| [Warnings and precautions]
Gefitinib is not suitable for pregnant women, and breastfeeding women should cease breastfeeding throughout their treatment period.
|
|
|