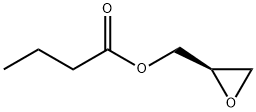
(R)-Glycidyl butyrate synthesis
- Product Name:(R)-Glycidyl butyrate
- CAS Number:60456-26-0
- Molecular formula:C7H12O3
- Molecular Weight:144.17
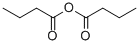
106-31-0
226 suppliers
$10.00/5g
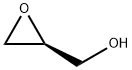
60456-23-7
246 suppliers
$8.00/1g
60456-26-0
308 suppliers
$14.14/1gm:
Yield: 98.1 - 99.5 % ee
Reaction Conditions:
with triethylamine;dmap in dichloromethane at 0 - 20; for 1 h;Product distribution / selectivity;
Steps:
1
To 1.2 L of a methylene chloride solution of (S)-3-chloro-l,2-propanediol (200 g, 99.5% ee) was added 519 g of potassium phosphate tribasic, and then the obtained solution was refluxed, under stirring, for 3 hours. The resulting solution was cooled to 0°C, and 220 g of trie thy lamine, 4 g of 4-(dimethylamino)pyridine, and 315 g of butanoic acid anhydride were dropwisely added to the solution. After additional stirring for 1 hour at a room temperature, the reaction mixture was successively washed with 2.2 L of 5% aqueous potassium carbonate solution, 2 L of IN aqueous hydrogen chloride solution, and 1 L of water. The organic layer was dried with 50 g of anhydrous sodium sulfate and filtrated. The methylene chloride was evaporated under reduced pressure. Fractional distillation (90°C/19 mmHg) of the resulting residue gave 242 g of the targeted compound:- Yield: 92.7%- Chemical purity: 99.4%- Optical purity (GC) 99.5% ee Comparative Example 1 Preparation of (R)-glycidyl butyrateTo 1.2 L of a methylene chloride solution of (S)-3-chloro-l,2-propanediol (200 g, 99.5% ee) was added 338 g of potassium carbonate, and then the obtained solution was refluxed, under stirring, for 25 hours. The resulting solution was cooled to 0°C, and 220 g of triethylamine, 4 g of 4-(dimethylamino)pyridine, and 315 g of butanoic acid anhydride were dropwisely added to the solution. After additional stirring for 1 hour at a room temperature, the reaction mixture was successively washed with 2.2 L of 5% aqueous potassium carbonate solution, 2 L of IN aqueous hydrogen chloride solution, and 1 L of water. The organic layer was dried with 50 g of anhydrous sodium sulfate and filtrated. The methylene chloride was evaporated under reduced pressure. Fractional distillation (90°C/19 mmHg) of the resulting residue gave ID g of the targeted compound:- Yield: 65.0%- Chemical purity: 97.4%- Optical purity (GC) 98.1% eeAs shown in the above, the process according to the present invention provides glycidyl derivatives in high yield, with high chemical purity and high optical purity, compared to the conventional one in which the carbonate salt was used. In addition, the reaction rate of the phosphate salt is even faster than that of the carbonate salt. Therefore, the process according to the present invention is an industrially ad-vantageous one. Furthermore, the process according to the present invention can be applicable to the preparation of various glycidyl derivatives including a chiral glycidyl alkanoate as well as a chiral glycidyl sulfonate.
References:
RSTECH CORPORATION WO2006/19202, 2006, A1 Location in patent:Page/Page column 6-7; 9
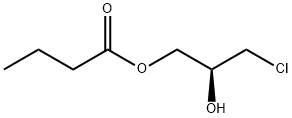
890051-54-4
0 suppliers
inquiry
60456-26-0
308 suppliers
$14.14/1gm:
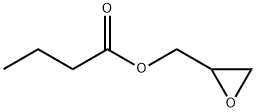
2461-40-7
42 suppliers
inquiry
60456-26-0
308 suppliers
$14.14/1gm:
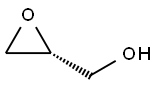
57044-25-4
252 suppliers
$6.00/1g

141-75-3
342 suppliers
$12.32/25G
60456-23-7
246 suppliers
$8.00/1g
60456-26-0
308 suppliers
$14.14/1gm:
2461-40-7
42 suppliers
inquiry
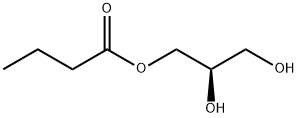
5309-42-2
3 suppliers
inquiry
60456-26-0
308 suppliers
$14.14/1gm:
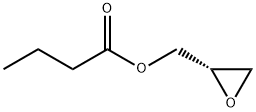
65031-96-1
199 suppliers
$16.00/5g