| 99% |
With hydrogen;palladium 10% on activated carbon; In ethanol;Inert atmosphere of nitrogen; |
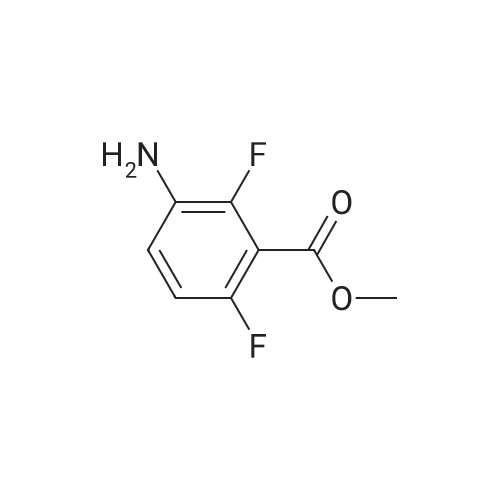
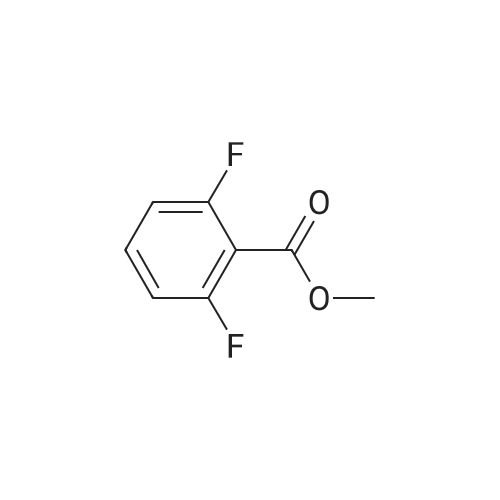
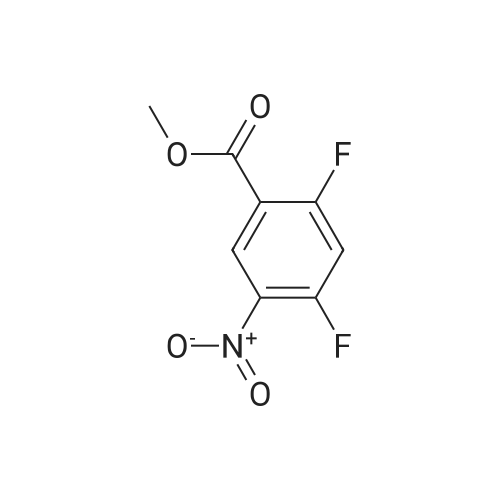
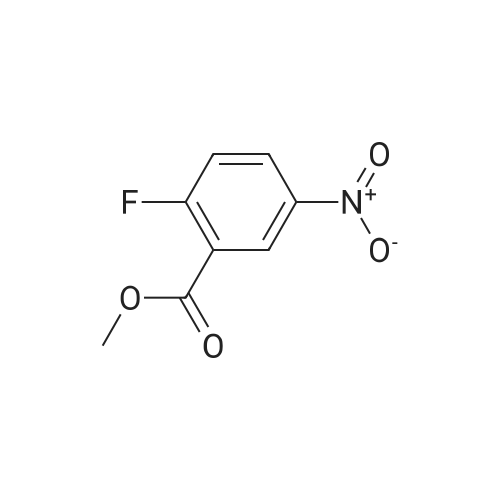
10% (wt.) Pd on activated carbon (4.46 g, 4.19 mmol) was added to a1 L flask charged with methyl 2,6-difluoro-3-nitrobenzoate (18.2 g, 83.8 mmol) under a nitrogen atmosphere. EtOH (350 mL, 0.25 M) was added, and then H2 was passed through the reaction mixture for 15 minutes. The reaction mixture was stirred under two H2 balloons overnight. The following day the reaction mixture was re-flushed with fresh H2 balloons and stirred an additional 4 hours. Upon consumption of the starting material and intermediate hydroxylamine as determined by TLC, N2 gas was flushed through the reaction mixture. The <n="68"/>mixture was then filtered through glass microfibre filter ("GF/F") paper twice. The volatiles were removed to afford methyl 3-amino-2,6-difluorobenzoate as an oil (15.66 g, 99%). The material was taken directly onto the next step. |
| 98% |
With palladium on activated charcoal; hydrogen; In ethanol; at 20℃; |
General procedure: Pd/C (0.1 eq.) was added to a solution of the nitrobenzene (1.0 eq.) in EtOH (0.2 m). The suspension was degassed with H2and the reaction was stirred at room temperature upon complete consumption of the starting material. Then, the mixture was passed through a Celite pad and the filtrate was concentrated in vacuum. The product was used without any further purification. |
| 98% |
With hydrogenchloride; iron; In ethanol; water; at 80℃; |
Alternatively: 75 (1 g, 4.6 mmol, 1.0 eq.) was suspended in abs.Ethanol (0,25 M) and aqueous HCl solution (1 M, 4.6 ml, 1.0 eq.) andheated to 80 C. Fe0 (282 mg, 5.1 mmol, 1.1 eq.) was added to themixture and stirred at 80 C until complete consumption of the starting material. The crudewas poured through a pad of Celite andthe filtrate dried in vacuo. The productwas obtained as a white solidand was used without further purification steps. Yield: 843 mg,4.5 mmol, 98%. |
| 93% |
With palladium 10% on activated carbon; hydrogen; In ethanol; under 2585.81 Torr; for 22h; |
A solution of compound 2 (15.0 g, 69.1 mmol) in EtOH (250 mL) was hydrogenation over 10% Pd/C (4.0 g) at 50 psi for 22 h. The catalyst was filtered off through a layer of Celite, and the filtrate was concentrated in vacuo. The crude product was purified by silica gel column chromatography (4:1-3:1 hexanes/EtOAc) to give 3 (12.9 g, 93%) as a pale yellow oil. 1H NMR (DMSO-d6): δ 6.93-6.91 (m, 2H), 5.26 (br s, 2H), 3.87 (s, 3H) |
| 93% |
With hydrogen;5%-palladium/activated carbon; In methanol; at 25℃; under 2585.81 Torr; for 12h;Inert atmosphere; |
To a solution of methyl 2,6-difluoro-3-nitrobenzoate (25 g, 1 15 mmol) in MeOH (150 ml_) was added 5% palladium on carbon (2.5 g). The mixture was stirred under H2 atmosphere (50 psi/25 C) for 12 h. The reaction mixture was filtered, and the filtrate was concentrated under the reduced pressure to give the product. 20 g (93 % yield) 1 H NMR (400 MHz, DMSO-c/6) δ ppm 6.95-7.10 (m, 2H), 3.86 (s, 3H) |
| 91% |
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; |
Step 4: methyl 3-amino-2,6-difluorobenzoateTo a solution of methyl 2,6-difluoro-3-nitrobenzoate (50 g, 0.23 mol) in MeOH (150 mL) was added Pd/C (10%) and the resulting reaction mixture was stirred at room temperature for overnight under H2 atmosphere. The mixture was filtered. The filtrate was concentratd in vacuo to afford the desired product (38.8 g, 91 >).1H NMR (CDCI3): ? 6.84-6.73 (2H, m), 3.94 (3H, s), 3.69 (2H, br). |
| 91% |
With palladium on activated charcoal; hydrogen; In methanol; at 20℃; |
To a solution of 2,6-difluoro-3-nitrobenzoate (50 g, 0.23mo 1) in methanol was added palladium on carbon (10%).The resulting mixture was stirred overnight at room temperature under a hydrogen atmosphere, filtered, and the resulting filtrate was concentrated in vacuo to give the title compound (38.8 g, 91%) |
|
With hydrogen;palladium 10% on activated carbon; In ethanol;Inert atmosphere; |
Step B: 10% (wt.) Pd on activated carbon (4.46 g, 4.19 mmol) was added to a 1 L flask charged with methyl 2,6-difluoro-3-nitrobenzoate (18.2 g, 83.8 mmol) under a nitrogen atmosphere. To the flask was added EtOH (350 mL, 0.25 M), and H2 gas was passed through the mixture for 15 minutes. The reaction mixture was stirred under two H2 balloons overnight. The balloons were recharged with H2 gas and the mixture was stirred an additional 4 hours. Upon consumption of the starting material and intermediate hydroxylamine as determined by TLC, N2 gas was flushed through the reaction mixture. The mixture was then filtered through glass microfibre filter ("GF/F") paper twice. The volatiles were removed to afford crude methyl 3-amino-2,6-difluorobenzoate as an oil (15.66 g). The material was taken directly onto the next step. |
|
With hydrogen;palladium 10% on activated carbon; In ethanol; |
10% (wt.) Pd on activated carbon (4.46 g, 4.19 mmol) was added to a 1 L flask charged with methyl 2,6-difluoro-3-nitrobenzoate (18.2 g, 83.8 mmol) under a nitrogen atmosphere. EtOH (350 mL, 0.25 M) was added, and then H2 was passed through the reaction mixture for 15 minutes. The reaction mixture was stirred under two H2 balloons overnight. The following day the reaction mixture was re-flushed with fresh H2 balloons and stirred an additional 4 hours. Upon consumption of the starting material and intermediate hydroxylamine as determined by TLC, N2 gas was flushed through the reaction mixture. The mixture was then filtered through glass Dulfonamid filter ("GF/F") paper twice. The volatiles were removed to afford methyl 3-amino-2,6-difluorobenzoate as an oil (15.66 g). The material was taken directly onto the next step. |
|
With hydrogen;palladium 10% on activated carbon; In ethanol; |
10% (wt.) Pd on activated carbon (4.46 g, 4.19 mmol) was added to a 1 L flask charged with methyl 2,6-difluoro-3-nitrobenzoate (18.2 g, 83.8 mmol) under a nitrogen atmosphere. EtOH (350 mL, 0.25 M) was added, and then H2 was passed through the reaction mixture for 15 minutes. The reaction mixture was stirred under two H2 balloons overnight. The following day the reaction mixture was re-flushed with fresh H2 balloons and stirred an additional 4 hours. Upon consumption of the starting material and intermediate hydroxylamine as determined by TLC, N2 gas was flushed through the reaction mixture. The mixture was then filtered through glass microfibre filter ("GF/F") paper twice. The volatiles were removed to afford methyl 3-amino-2,6-difluorobenzoate as an oil (15.66 g). The material was taken directly onto the next step. |
|
With hydrogen;palladium 10% on activated carbon; In ethanol;Inert atmosphere; |
Step B: 10% (wt.) Pd on activated carbon (4.46 g, 4.19 mmol) was added to a 1L flask charged with methyl 2,6-difluoro-3-nitrobenzoate (18.2 g, 83.8 mmol) under a nitrogen atmosphere. To the flask was added EtOH (350 mL, 0.25 M), and H2 gas was passed through the mixture for 15 minutes. The reaction mixture was stirred under two H2 balloons overnight. The balloons were recharged with H2 gas and the mixture was stirred an additional 4 hours. Upon consumption of the starting material and intermediate hydroxylamine as determined by TLC, N2 gas was flushed through the reaction mixture. The mixture was then filtered through glass microfibre filter ("GF/F") paper twice. The volatiles were removed to afford crude methyl 3-amino-2,6-difiuorobenzoate as an oil (15.66 g). The material was taken directly onto the next step. |
|
With hydrogen;palladium 10% on activated carbon; In ethanol;Inert atmosphere; |
10% (wt.) Pd on activated carbon (4.46 g, 4.19 mmol) was added to a 1 L flask charged with methyl 2,6-difluoro-3-nitrobenzoate (18.2 g, 83.8 mmol) under a nitrogen atmosphere. To the flask was added EtOH (350 mL, 0.25 M), and H2 gas was passed through the mixture for 15 minutes. The reaction mixture was stirred under two H2 balloons overnight. The balloons were recharged with H2 gas and the mixture was stirred an additional 4 hours. Upon consumption of the starting material and intermediate hydroxylamine as determined by TLC, N2 gas was flushed through the reaction mixture. The mixture was then filtered through glass microfibre filter ("GF/F") paper twice. The volatiles were removed to afford crude methyl 3-amino-2,6-difluorobenzoate as an oil (15.66 g). The material was taken directly onto the next step. |
|
With iron; acetic acid; In methanol; water; at 110℃; |
Iron powder (15.5g, 4.0 equiv.) was added to 100 mL of water, and 100 mL of acetic acid was added under stirring, followed by 2,6-difluoro-The 3-nitrobenzoic acid methyl ester compound 2309 (15.0 g, 0.069 mol) was dissolved in ethanol (100 mL) and added with a small amount of acetic acid to aid dissolution.The mixture was slowly added to the above mixed solution and heated to 110C. TLC showed that the reaction was complete. The reaction solution was distilled off under reduced pressure and the residue remainedEthyl acetate was added to the solution, which was filtered. The filtrate was washed with saturated sodium bicarbonate, washed with water, saturated brine, and dried over anhydrous sodium sulfate.This was evaporated to dryness to afford compound 2310 (13.2 g, yield: 100%) which was used in the next step without purification |
|
With palladium 10% on activated carbon; hydrogen; In ethanol; |
Methyl 2,6-difluoro-3-nitrobenzoate (1.13 g, 5.19 mmol) was dissolved in ethanol (17.0 ml.) and palladium on activated charcoal (10% Pd) (0.055g, 0.052 mmol) was added while stirring. The reaction was flushed with hydrogen until complete consumption of the starting material. The suspension was filtrated and the solvent of the filtrate was removed under vacuum. The residue was purified via flash chromatography (silica, gradient: 100% n-hexane -> n- hexane/ethyl acetate 6/4 v/v). The product was obtained as yellow oil. 1H-NMR (200 MHz, DMSO-de) d 7.00 - 6.82 (m, 2H), 5.26 (s, 2H), 3.86 (s, 3H). 13C-NMR (50 MHz, DMSO- d6) d 162.0, 149.8 (dd, J = 240.7, 5.4 Hz), 146.6 (dd, J = 247.6, 6.5 Hz), 133.7 (dd, J = 12.9, (0255) 2.6 Hz), 1 18.2 (dd, J = 8.9, 6.8 Hz), 1 11.6 (dd, J = 22.0, 3.6 Hz), 1 10.1 (dd, J = 20.0, 16.4 Hz), 52.8. TLC-MS (ESI-): Calculated 187.04 for C8H7F2NO2. Measured 185.6 for [M-H] . HPLC: tR = 3.980 min, purity: 97.4% (254.4 nm), 97.6% (230.4 nm). |
|
With palladium 10% on activated carbon; hydrogen; In ethanol; |
To a 1 L flask charged with methyl 2,6-difluoro-3-nitrobenzoate (18.2 g, 83.8 mmol) was added 10% wt. Pd on activated carbon (4.46 g, 4.19 mmol) under a nitrogen atmosphere. To the flask was added EtOH (350 mL), and hydrogen was passed through the mixture for 15 minutes. The reaction mixture was stirred under two hydrogen balloons overnight. The balloons were recharged with H2 (g) and the mixture was stirred an additional 4 hours. Upon consumption of the starting material and intermediate hydroxylamine as determined by TLC, nitrogen was flushed through the reaction mixture. The mixture was filtered through glass microfibre filter (GF/F) paper twice. The solution was concentrated to afford crude methyl 3-amino-2,6-difluorobenzoate as an oil (15.66 g). The material was used without further purification. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping