| 99% |
With sodium hydroxide; hydroxylamine hydrochloride; In ethanol; water; at 90℃;Product distribution / selectivity; |
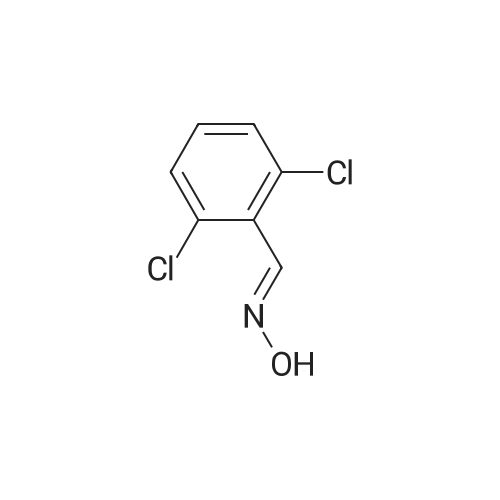
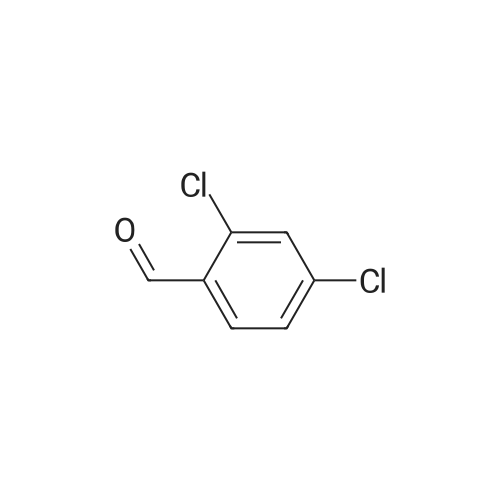
Example 32; 6-{4-[5-Cvclopropyl-3-(2,6-dichloro-phenyl)-isoxazol-4-ylmethoxy1-piperidin-l-yl}-l- methyl-1 H-indole-3 -carboxylic acid; <n="56"/>Step l; 2.6-Dichloro-benzaldehyde oxime; Sodium hydroxide 3N (3.14L, 9.43mol) is added dropwise to a stirred suspension of hydroxylamine hydrochloride (675.55g, 9.43mol) in 0.5L of water at O0C. To this mixture is added dropwise a suspension of 2,6-dichlorobenzaldehyde (150Og, 8.57mol) in 7.5L of ethanol and the reaction is heated at 9O0C overnight. The mixture is cooled to room temperature and then, concentrated to dryness. The solid is triturated in a mixture of H2O/EtOH, 10: 1 (4.4L), filtered and dried under high vacuum at 450C overnight. 1621.78g of title compound (99% yield) is obtained as a white solid. MS (m/e): 190 (M+ 1) |
| 98% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 0 - 90℃; for 24h; |
To a solution of NH2OH·HCl (10.9 g, 0.157 mol) in water (100 mL) was added NaOH (6.27 g, 0.157 mol) at 0 C. The resulting solution was then added to a solution of 1 (25.0 g, 0.142 mol) in ethanol (200 mL). The resulting mixture was stirred at 90C for 24 h. The reaction mixture was concentrated under reduced pressure. The solids were collected by filtration and washed with water to give 2 (26.5 g, 98%) as a white compound which was used in the next step without further purification. |
| 98% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 90℃; |
Hydroxylamine hydrochloride (67.6 g, 0.943 mol) was dissolved in 100 mL of water, and sodium hydroxide 3N was added dropwise under stirring at 0 C.(314 mL, 9.43 mol. 2,6-dichlorobenzaldehyde (150 g, 0.857 mol) was added to ethanol (300 mL) to dissolve, and the mixture solution was added dropwise, and then the temperature was raised to 90 C. and stirred overnight.The reaction was monitored by thin layer chromatography (TLC). After the reaction was completed, it was cooled to room temperature and concentrated in vacuo to a 10% solution.The solid was obtained by suction filtration and dried to obtain 162 g of a white solid.Yield: 98%. |
| 97% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 0 - 90℃; |
To a 2 L round-bottom flask containing hydroxylamine hydrochloride (108 g, 1.55mol, 1.3 equiv.), sodium hydroxide (60 g, 1.50 mol, 1.3 equiv.), and water (200 mL) was5 added 2,6-Dichlorobenzaldehyde 1a (200 g, 1.14 mol, 1.0 equiv.) dropwise at 0 C, folov,'edby ethanol (500 mL). The resulting rnixture was heated at 90 C ovemight, and thenconcentrated under reduced pressure. The resulting solids were collected by filtration anddried in an oven tmder reduced pressure, to provide 210 g (97%) of N-[ (2,6-dichlorophenyl)methylidene]-hydroxy lamine l bas an ofi-white solid. |
| 96% |
With sodium hydroxide; hydroxylamine hydrochloride; In ethanol; water; at 90℃; for 24h; |
A solution of 2, 6-DICHLOROBENZA . DEHYDE (25g, 0.14 mole) in ethanol (200 mL) was added to a solution of HYDROXYLAMINE HYDROCHLORIDE (11G, 0.16 mole) and sodium hydroxide (6.3g, 0.16 mole) in water (100 mL). The resulting mixture was stirred at 90 C for 24 hours. The volume was reduced in vacuo by ca 30 mL which induced a precipitate. The flask was then cooled to room temperature and the white solids were collected by filtration and washed with water (2x 100 ML). Yield = 25.9g. (96%) of 2, 6-DICHLOROBENZALDEHYDE oxime. |
| 96% |
With sodium hydroxide; hydroxylamine hydrochloride; In ethanol; water; at 90℃; for 24h; |
A solution of 2,6-dichlorobenzaldehyde (25 g, 0.14 mole) in ethanol (200 mL) was added to a solution of hydroxylamine hydrochloride (11 g, 0.16 mole) and sodium hydroxide (6.3 g, 0.16 mole) in water (100 mL). The resulting mixture was stirred at 90° C. for 24 hours. The volume was reduced in vacuo by ca 30 mL which induced a precipitate. The flask was then cooled to room temperature and the white solids were collected by filtration and washed with water (2 100 mL). Yield=25.9 g. (96%) of 2,6-dichlorobenzaldehyde oxime. A 500 mL round bottom flask was charged with a solution of 2,6-dichlorobenzaldehyde oxime (13 g, 0.07 mole) in N,N-dimethyl formamide (150 mL). The flask was placed in an ambient temperature water bath. The flask was then charged with N-chlorosuccinimide (9.2 g, 0.07 mole). Within minutes of dissolution, an exotherm was observed along with a significant color change to dark yellow. The reaction was stirred an additional hour then the contents were then poured into water (200 mL) and the product extracted with diethyl ether (300 mL). The ethereal layer was washed with water (3 100 mL) and brine (50 mL), then dried over anhydrous magnesium sulfate. After filtering, the solvent was removed in vacuo to yield 14.5 g of a yellow oil. (94%) of 2,6-dichlorophenyl hydroximic chloride which was used without further purification. A stirred solution of methyl isobutyryl acetate (2 g, 15.6 mmol) in tetrahydrofuran (15 mL) was treated with a solution of sodium methoxide (31.5 mL, 0.5 M in methanol) followed by a solution of 2,6-dichlorophenyl hydroximic chloride (3.5 g, 15.6 mmol) in tetrahydrofuran (5 mL). After stirring at ambient temp 16 h the solvent was removed in vacuo. The resulting residue was partitioned with diethyl ether (100 mL) and water (100 mL). The ethereal layer was washed with brine (50 mL), dried over anhydrous magnesium sulfate, filtered and condensed to an oil. The product was purified by flash chromatography on silica gel using 10% ethyl acetate in hexane as mobile phase. Yield=3.1 g. (62%) of 3-(2,6-dichlorophenyl)-4-carbomethoxy-5-isopropyl-isoxazole. |
| 96% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 90℃; for 24h; |
A solution of 2,6-dichlorobenzaldehyde A1a (25 g, 0.14 mol) in 200 mL of ethanol was added to a solution of hydroxylamine hydrochloride (11 g, 0.16 mol) and sodium hydroxide (6.3 g, 0.16 mol) in 100 mL of water. The resulting mixture was stirred at 90C for 24 h. The volume was reduced in vacuum by - 30 mL, which induced a precipitate. The flask was then cooled to room temperature and the solid was collected by filtration and washed with water (2 x 100 mL). The solid was dried under vacuum to give 25.9 g of compound A2a (white solid, yield: 96%). |
| 96.5% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 90℃; |
A 3 mol / L sodium hydroxide solution (21 ml, 0.063 mol, 1.1 eq) was added dropwise to a suspension of 3.3 ml of water in hydroxylamine hydrochloride (4.37 g, 0.063 mol, 1.1 eq). The mixed solution was poured into a mixed solution of 50 ml of ethanol and 2,6-dichlorobenzaldehyde (10 g, 0.057 mol, 1.0 eq), and the mixture was heated to 90 C overnight. The reaction solution was concentratedDry, concentrated dry solid was added 29.3 ml (H2O: EtOH = 10: 1) solution, filtered and crystallized,The filter cake was dried and dried in vacuo at 45 C to give intermediate 1-8, 10.48 g as a white solid, 96.5% yield. |
| 96.5% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 90℃; |
At 0 C, 3 mol/L sodium hydroxide solution (21 ml, 0.063 mol, 1.1 eq) was dropped into suspended 3.3 ml waterA solution of hydroxylamine hydrochloride (4.37 g, 0.063 mol, 1.1 eq).The mixed solution was added dropwise to a mixed solution of 50 ml of ethanol and 2,6-dichlorobenzaldehyde (10 g, 0.057 mol, 1.0 eq), and the mixture was heated to 90 C overnight.After the completion of the reaction, the reaction solution was concentrated to dryness, and then concentrated, dried solid was added to a solution of 29.3 ml (H2O:EtOH=10:1).After beating and crystallization, the filter cake was drained and dried under vacuum at 45 C.Intermediate 1-8 was obtained as a white solid 10.48 g, yield 96.5%. |
| 96.5% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 90℃; for 24h; |
Hydroxylamine hydrochloride (10.9g) and sodium hydroxide (6.27g) were dissolved in water at room temperature.Add dropwise to a solution of 2,6-dichlorobenzaldehyde (25 g) in ethanol (200 mL).Stir at 90 degrees for 24 hours.Cool to room temperature,Spin dry ethanol,filter,Wash the filter cake with water,The product was dried under infrared light to obtain 26.5 g of a product.The yield was 96.5%. |
| 96% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 90℃; for 24h; |
Sodium hydroxide(6.3g, 160mmol) and 2,6-dichlorobenzaldehyde(25g, 140mmol) in ethanol(200ml) was added to hydroxylamine hydrochloride(11g, 160mmol) in water(100ml) and stirred for 24 hours at 90 . The reaction mixture was evaporated in vacuum, filtered with water(200ml, 2 times) and dried in vacuum to afford the intermediate compound 2,6-dichlorobenzaldehyde oxime(25.9g, 96%). [585] 1H-NMR (DMSO, 400MHz): δ 11.80 (s, 1H), 8.22 (s, 1H), 7.55 (d, 2H), 7.45-7.41 (dd, 1H). |
| 96% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 90℃; for 24h; |
Hydroxylamine hydrochloride (11 g, 160 mmol) was added to a solution of to distilled water (100 ml) and sodium hydroxide (6.3 g, 160 mmol) and 2,6-Dichlorobenzoaldehyde (25 g, 140 mmol) was added to a solution of Dissolved in ethanol (200 ml) was added, and the mixture was stirred at 90 C for 24 hours.The reaction mixture was concentrated and the resulting solid was washed with distilled water (200 ml, twice) and dried to give the intermediate compound 2,6-dichlorobenzaldehyde oxime (25.9 g, 96%) was obtained. |
| 94% |
With hydroxylamine hydrochloride; triethylamine; In dichloromethane; for 8h;Product distribution / selectivity; |
Alternate procedure: Add triethylamine (23.1 g, 229 mmol) dropwise to a solution of 2,6-dichloro-benzaldehyde (20.0 g, 114 mmol) and hydroxylamine hydrochloride (10.3 g, 149 mmol) in dichloromethane (200 mL). Stir the reaction mixture for 8 h. Add water (200 mL). Separate the phases and extract the aqueous phase with dichloromethane (100 mL). Wash the combined organic phases with water (100 mL). Concentrate the combined organic phases to provide 28.8 g (94%) of the title compound. |
| 89% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 90℃; |
2,6-dichlorobenzaldehyde (611) (25g,0.14mol) was dissolved in 200mL ethanol, Followed by addition hydroxylamine hydrochloride (0.16 mol)And 6.3 g of sodium hydroxide (0.16 mol) in 100 mL of an aqueous solution.Then the system temperature to 90 C,After the reaction at this temperature for 2-3 h, TLC detection reaction.After the reaction, The solvent was evaporated to about 30 mL, and a large amount of solid was precipitated. The solid was washed with water and dried to give 24 g of product 2,6-dichlorobenzyloxime (AI) (89%). |
| 85% |
With pyridine; hydroxylamine hydrochloride; In ethanol; at 0℃;Reflux; |
2,6-dichlorobenzaldehyde (25 gm, 0.143 moles, 1 eq) was dissolved in EtOH (200 ml) followed by the addition of hydroxyl amine hydrochloride (19.9 gm, 0.286 moles, 2 eq) and pyridine (34 gm, 0.429 moles, 3 eq) at 00C. The reaction mixture was refluxed for 40 minutes and checked by TLC. The solvent was removed under reduced pressure and to the residue water was added and extracted with ethyl acetate twice. The combined organic layer was washed with brine, dried over Na2SO4, concentrated and washed with chilled hexane to afford the mixture of syn and anti oxime b in 85% yield (23 gm). MS 190.0 (M+ peak) 1H NMR (200 MHz, CDCl3): δ 7.20-7.30 (m, 3H), 7.38-4.00 (m, 3H), 8.20 (s, IH), 8.40 (s, IH) mixture of syn and anti isomers. |
| 85% |
With pyridine; hydroxylamine hydrochloride; In ethanol; at 0℃; for 0.666667h;Reflux; |
2,6-dichlorobenzaldehyde (25 gm, 0.143 moles, 1 eq) was dissolved in EtOH (200 ml) followed by the addition of hydroxyl amine hydrochloride (19.9 gm, 0.286 moles, 2 eq) and pyridine (34 gm, 0.429 moles, 3 eq) at 00C. The reaction mixture was refluxed for 40 minutes and checked by TLC. The solvent was removed under reduced pressure and to the residue water was added and extracted with ethyl acetate twice. The combined organic layer was washed with brine, dried over Na2SO4, concentrated and washed with chilled hexane to afford the mixture of syn and anti oxime b in 85% yield (23 gm). MS 190.0 (M+ peak) 1H NMR (200 MHz, CDCl3): δ 7.20-7.30 (m, 3H), 7.38-4.00 (m, 3H), 8.20 (s, IH), 8.40 (s, IH) mixture of syn and anti isomers. |
| 78% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 90℃; for 16h; |
3N Sodium hydroxide (8.35 g, 0.208 mol) was added drop wise to a solution of hydroxylamine hydrochloride (l4.5lg, 0.208 mol) in water (130 ml) at 0 C . A solution of 2, 6-dichloro-benzaldehyde (32.0g, 0.182 mol) in ethanol (250 ml) was then added and the reaction mixture heated at 90 C for 16 h. The mixture was then cooled to room temperature, concentrated to dryness and the crude product triturated with 10:1 water/EtOH, filtered and dried under reduced pressure to afford the titled compound as a solid (27.0 g, 78 % yield). 'H NMR (400 MHz, d6-DMSO): d 11.79 (s, 1H), 8.22 (s, 1H), 7.60-7.38 (m, 3H). |
| 64% |
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; at 90℃; |
2,6-Dichlorobenzaldehyde (5.0 g, 28.6 mmol) is dissolved in anhydrous ethanol (45 ml), and the mixture is stirred at room temperature, followed by adding NH2OH·HCl (2.3 g, 33.1 mmol), NaOH (1.3 g, 32.5 mmol) and water (20 mL) with overnight reflux in an oil bath at 90 C.When the reaction mixture is cooled to room temperature, the mixture is rotated to remove solvents, followed by adding water (100 mL), and then it is extracted with ethyl acetate (2 * 100 mL), then the organic solution is combined and washed with saturated salt water (2 * 100 mL), and then is dried with anhydrous Na2SO4, then filtered and rotated to dryness, after that, adding petroleum ether (100 mL), and stirring well, finally the product is filtered and dried, so that a white solid, that is 2,6-dichlorobenzaldehyde oxiame, is obtained with a yield of 64%. |
|
With sodium hydroxide; water; In methanol; |
Step 1 2.6-Dichloro-bcnzaldchvdc oximc2,6-Dichloro-benzaldelryde (7.0 g,40 mmol) is added to 10 mL of water and 30 mL of methanol. Sodium hydroxide (4.0 g, 100 mmol) is dissolved in 8 mL of water slowly. The sodium hydroxide solution <n="12"/>is added to the benzaldelαyde solution. The reaction is stirred overnight. The reaction mixture is partitioned between ethyl acetate and water. The organic layer is washed with brine and dried over solid sodium sulfate. The organic layer is filtered and the solvent is removed under reduced pressure to yield the title compound. |
|
With sodium hydroxide; hydroxylamine hydrochloride; In ethanol; water; at 20℃; for 1h; |
BY THE GENERAL PROCEDURE OF R. K. HOWE, ET AL, J. ORG. CHEM., 1980,45, 3916-3918 the aldehyde starting material 245 in 1: 1 ethanol-water was treated with 1.1 equivalents of hydroxylamine hydrochloride and 2.5 equivalents of aqueous sodium hydroxide with cooling. The mixture was then stirred at room temperature for LH. The reaction mixture was extracted with ether, which was discarded and the aqueous layer was separated and acidified to pH 6 with concentrated hydrochloric acid with cooling. The aqueous layer was extracted with ether and the ether layers were separated. The combined ether layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to yield the desired solid oximes. |
|
With pyridine; hydroxylamine hydrochloride; at 20℃; |
Referring to Fig. 3A, the aldehyde starting material 245 was dissolved in pyridine solvent, and 1.0-1. 2 equivalents of solid hydroxylamine hydrochloride was added in one portion and the homogeneous mixture was stirred overnight at room temperature. The mixture was concentrated under reduced pressure. The residue was dissolved in ethyl acetate and this solution was washed with either 1N hydrochloric acid followed by saturated brine, or by saturated brine alone. The ethyl acetate solution was then dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to yield the desired oxime, 247 |
|
With sodium hydroxide; hydroxylamine hydrochloride; In methanol; water;Product distribution / selectivity; |
Intermediate Preparation 2; 2,6-Dichloro-benzaldehyde oxime; 2,6-Dichloro-benzaldehyde (7.0 g, 40 mmol) and hydroxylamine hydrochloride (2.16 g, 44 mmol) are added to 10 mL of water and 30 mL of methanol. Sodium hydroxide (4.0 g, 100 mmol) is dissolved in 8 mL of water slowly. The sodium hydroxide solution is added to the benzaldehyde solution. The reaction is stirred overnight. The reaction mixture is partitioned between ethyl acetate and water. The organic layer is washed with brine and dried over solid sodium sulfate. The organic layer is filtered and the solvent is removed under reduced pressure to yield the title compound. |
|
With hydroxylamine hydrochloride; In water; at 20℃; for 0.5h;Green chemistry; |
General procedure: The aqueous solution of hydroxylamine hydrochloride (1.2 mmol) and aldehyde 2 (1 mmol) were stirred at room temperature for 30 min. After complete conversion of aldehyde to oxime, N-chlorosuccinamide (1.3 mmol) was added to the reaction mixture and was allowed to stir for 3 h. The clay-Cu(II)/NaN3 mixture (prepared by stirring 15 mol % clay-Cu catalyst and 7.5 mol % NaN3 in water until the color changes from brown to black) and phenyl acetylene 5a (1.3 mmol) was added and the reaction mixture was further stirred for another 3 h. After completion of reaction, the reaction mixture was filtered through Whatman filter paper, residue was washed with EtOAc. Organic layer was separated from filtrate and was dried over anhydrous sodium sulfate. Combined organic layers were concentrated in vacuo and crude reaction mixture was purified by silica gel (100-200) column chromatography using EtOAc: hexane as eluting solvent to get corresponding 3,5-disubstituted isoxazoles 1a-1o in 68-88% yield. |
|
With hydroxylamine hydrochloride; sodium hydroxide; In ethanol; water; at 0 - 90℃; |
To a 2 L round-bottom flask containing hydroxylamine hydrochloride (108 g, 1.55 mol, 1.3 equiv.), sodium hydroxide (60 g, 1.50 mol, 1.3 equiv.), and water (200 mL) was added 2,6- dichlorobenzaldehyde la (200 g, 1 .14 mol, 1.0 equiv,), followed by ethanol (500 mL) at 0 C. The resulting mixture was stirred at 90 C overnight and then concentrated in vacuo. The resulting solids were collected by filtration and dried in an oven under reduced pressure, providing N-[(2,6-dichlorophenyl)methylidene]-hydroxylamine lb (210 g, 97%) as an off-white solid. The product was carried onto the next step without further purification. |
|
With hydroxylamine hydrochloride; sodium hydroxide; In water; at 83℃; for 3h;pH < 9.5; |
A solution of hydroxylamine hydrochloride (0.25 M) was prepared and sufficient quantity of water was added in order to keep the pH of solution below 2.5 and temperature of the solution below 25C. To this solution NaOH was added in order to keep the temperature below 50C and pH of the solution below 9.5. To this warm solution was slowly added 2,6-dichlorobenzaldehyde by keeping temperature below 83C. When addition of aldehyde was over, crystalline product formation was observed within 2-3 minutes. Reaction progress was monitored by TLC. Reaction was completed within 3 hours. Reaction mass was filtered and was dried in oven to obtain crystalline 2,6-dichlorobenzaldehyde oxime of formula VIII. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping