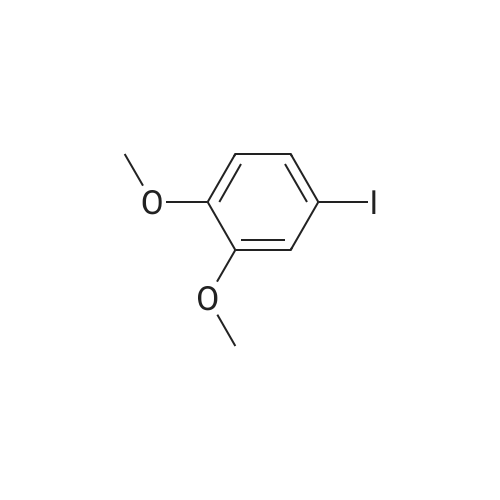
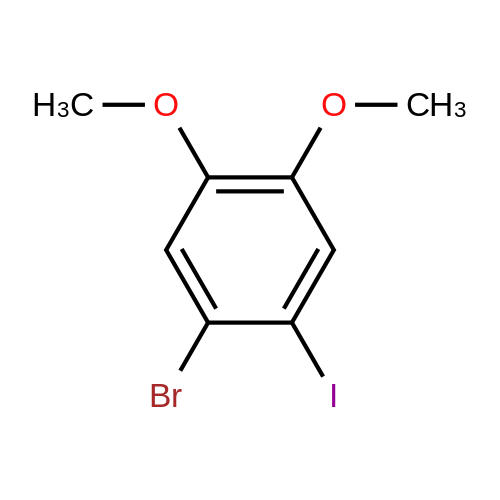
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
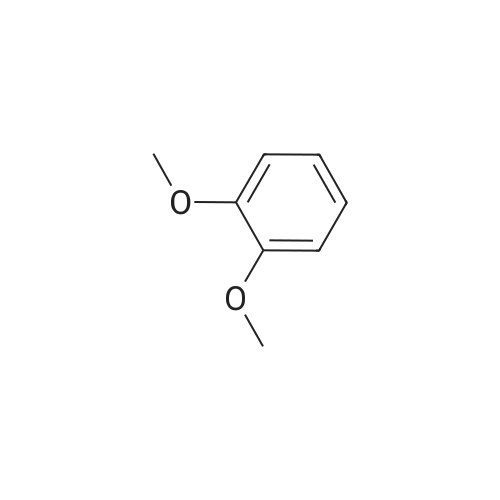
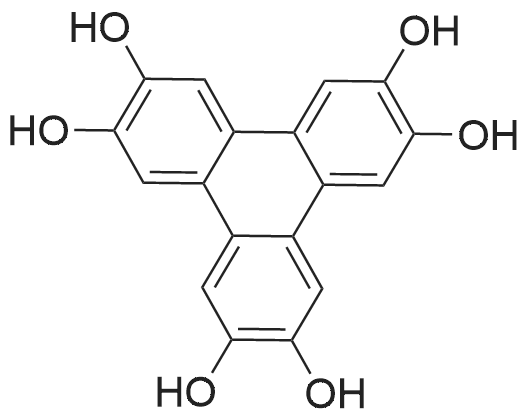
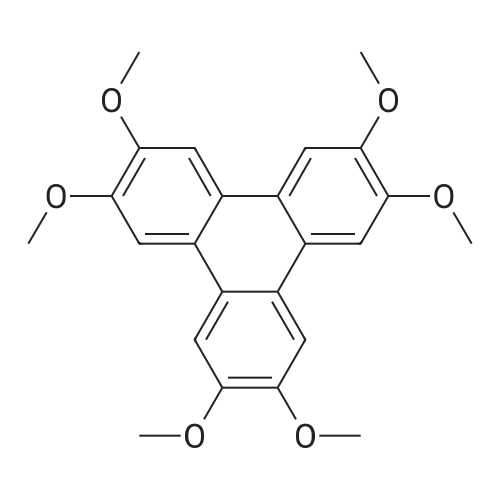
A type B crystal of 2,3,6,7,10,11-hexahydroxytriphenylene monohydrate was synthesized according to the process described in Synthesis, 477, 1994 and JP-A-8-119894. Namely, 1,2-dimethoxybenzene (31.78 g, 0.23 moles) and anhydrous ferric chloride (120 g, 0.74 moles) were dissolved in 70percent sulfuric acid, and the solution was reacted at 25°C for 24 hours with stirring. After completion of the reaction, the solution was poured into ice water (500 g), and the precipitated crystal was collected by filtration. After the resultant crystal was washed with water (1 L), and then dried to give pale purple colored 2,3,6,7,10,11-hexamethoxytriphenylene (28.2 g, theoretical yield from 1,2-dimethoxybenzene: 90.1percent) (the method of Synthesis, 477, 1994).
85.4%
at 10 - 15℃; for 6 h;
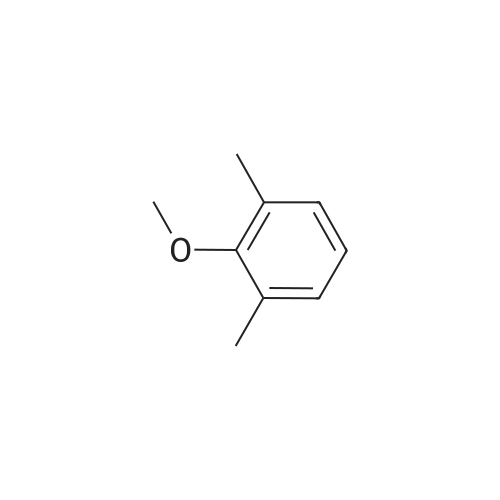
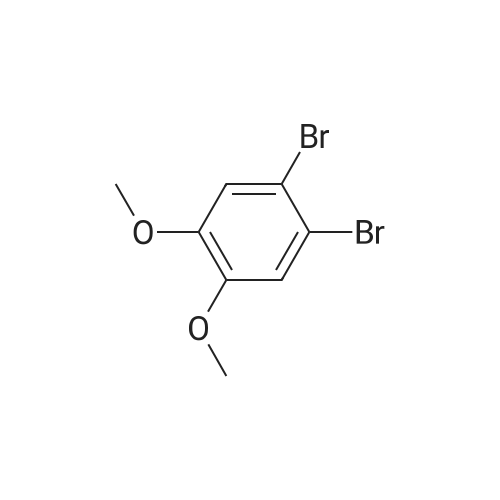
600 ml of ethyl acetate was added into a 1000ml four-necked flask, sodium peroxydisulfate 102.4g (0.43 mol) and o-xylene 45.7 g (0.43 mol) were added, stirred and cooled at internal temperature of 10 degrees C. Anhydrous iron(III) chloride 345.4g (2.10 mol) was added little by little, reacted at an internal temperature of 10-15 degrees C for 6 hours. After completion of the reaction, reaction mixture was cooled, 2000 ml of water was added and stirred for 10minutes. The aqueous layer of the solution was separated, and the organic layer was washed with 800 ml of salt solution. 600 ml of methanol was added to the organic layer and crystallized at 15-25 degrees C for 1 hour, the crystals were filtered, and dried to obtain 19.5 g (43.6percent of yield) of objects as gray crystal.
82%
With trifluorormethanesulfonic acid; 2,3-dicyano-5,6-dichloro-p-benzoquinone In 1,2-dichloro-ethane at 20℃; for 10 h; Inert atmosphere
General procedure: An oven-dried 20 mL scintillation vial, equipped with a magnetic stir-bar, was charged with the starting material (1.0 equiv), DDQ (1.0 equiv), trifluoromethanesulfonic acid (1.4percent v/v, 3.0 equiv), and 1,2-dichloroethane (0.05 M). The reaction mixture was then allowed to stir at ambient temperature for 10 h. After this time, methanol (0.05M) was added, and the solution was then allowed to stir at ambient temperature for an additional hour. Upon addition of the methanol, some solids precipitated out of the solution. Then, the solvent was removed from the heterogeneous mixture under reduced pressure. The crude material was purified by either recrystallization (methanol/DCM) or silica-gel column chromatography (hexanes/DCM) to give the title compounds.
80%
With iron(III) chloride; sulfuric acid In dichloromethane at 20℃; for 3 h;
A solution of 1,2-dimethoxybenzene (10 g, 72.4 mmol) in dichloromethane(50 ml) was added dropwise to a suspension of anhydrous FeCl3 (35.22 g, 217.2 mmol) in dichloromethane (100 ml) and concentrated sulphuric acid (0.5 ml). After complete addition (15 min), the reaction mixture was further stirred for 3 h at room temperature. 200 ml of methanol were then slowly added under vigorous stirring. The obtained mixture was further stirred for additional 30 min. And the precipitate was filtered off, washed with methanol (5 × 100 ml) and dried under reduced pressure to give a purple solid. Yield: 80percent, 1HNMR (CDCl3) δ/ppm: 4.10 (s, 18H, OCH3), 7.80 (s, 6H, ArH).
Reference:
[1] Angewandte Chemie - International Edition, 2010, vol. 49, # 44, p. 8209 - 8213
[2] Synthesis, 1994, vol. 1, # 5, p. 477 - 478
[3] Patent: EP2177495, 2010, A1, . Location in patent: Page/Page column 12
[4] Synthetic Communications, 1999, vol. 29, # 10, p. 1767 - 1771
[5] Journal of the American Chemical Society, 2017, vol. 139, # 46, p. 16759 - 16767
[6] Patent: JP5731346, 2015, B2, . Location in patent: Paragraph 0086; 0087; 0095
[7] Journal of Materials Chemistry, 2002, vol. 12, # 8, p. 2208 - 2213
[8] Tetrahedron Letters, 2015, vol. 56, # 23, p. 3458 - 3462
[9] Journal of Molecular Liquids, 2016, vol. 223, p. 734 - 740
[10] Journal of the American Chemical Society, 2009, vol. 131, p. 7662 - 7677
[11] Chemical Communications, 1997, # 17, p. 1615 - 1616
[12] Journal of the American Chemical Society, 1980, vol. 102, # 21, p. 6504 - 6512
[13] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1975, p. 2185 - 2189
[14] Tetrahedron, 1965, vol. 21, p. 3229 - 3236
[15] Tetrahedron, 1991, vol. 47, # 4/5, p. 791 - 798
[16] Molecular Crystals and Liquid Crystals (1969-1991), 1985, vol. 125, p. 279 - 288
[17] Journal of Structural Chemistry, 2001, vol. 42, # 1, p. 38 - 42
[18] Chemistry Letters, 1994, # 6, p. 981 - 984
[19] Journal of Physical Chemistry, 1995, vol. 99, # 3, p. 1005 - 1017
[20] Synthesis, 1997, # 11, p. 1285 - 1290
[21] Russian Chemical Bulletin, 2004, vol. 53, # 8, p. 1743 - 1748
[22] Chemical Communications, 2015, vol. 51, # 21, p. 4368 - 4371
[23] Chimia, 2015, vol. 69, # 9, p. 520 - 523
[24] Patent: JP2017/31106, 2017, A, . Location in patent: Paragraph 0052-0055
[25] Journal of Materials Chemistry A, 2017, vol. 5, # 24, p. 12080 - 12085
With boron tribromide; In dichloromethane; at 20℃;
The preparation of compound 4 is similar to that reported in the literature[14]. Hexamethoxytriphenylene (7 g, 17.14 mmol) was dissolved in dichoromethane (50 ml) and the solution thus obtained was cooled in ice bath. A solution of boron tribromide (11.47 ml) was then added slowly to the reaction mixture over a period of 30 min. After complete addition, the reaction was left stirring overnight in room temperature.The reaction mixture was then slowly poured into crushed ice(100 g) and the obtained mixture was stirred vigorously until the ice melted. It was then extracted with ethyl acetate (6 × 150 ml), dried over anhydrous sodium sulphate and evaporated to dryness giving dark purple solid. The yield was quantitative and this compound was used without further purification.
90%
With hydrogenchloride; acetic acid; In (2S)-N-methyl-1-phenylpropan-2-amine hydrate;
EXAMPLE 6 In a 50 cc glass sealed tube, 3.6 g of 2,3,6,7,10,11-hexamethoxytriphenylene, 20 ml of 35percent hydrochloric acid and 20 ml of acetic acid were placed. In an oven-type microwave irradiator where power and temperature are controllable, the mixture was continuously irradiated with microwaves having a frequency of 2.45 GHz and a power of 800 W, and then subjected to the reaction at 140° C. and 0.51 MPa for 2 hours. The reaction mixture was analyzed using liquid chromatography, showing a yield of the objective compound (2,3,6,7,10,11-hexahydroxytriphenylene) of 90.8percent and a residual ratio of the starting material (2,3,6,7,10,11-hexamethoxytriphenylene) of 0.0percent. Pouring this mixture into 100 ml of ice water produces the objective 2,3,6,7,10,11-hexahydroxytriphenylene in the form of crystal (purity:99percent, yield:90percent). 1H-NMR(DMSO-d6) delta ppm: 7.605(brs, 6H), 9.292(brs, 6H) 13C-NMR(DMSO-d6) delta ppm: 139.86, 153.96, 177.46
76%
With boron tribromide; In dichloromethane; at -70 - 25℃; for 10h;Inert atmosphere;
2,3,6,7,10,11-Hexamethoxytriphenylene (5.0 g, 12.2 mmol) was dissolved in 80 mL of dichloromethane under nitrogen and the reaction mixture was cooled to -70 °C. A solution of BBr3 (1M, CH2Cl2, 100 mL) was added dropwise to the above solution within a period of half an hour. The solution was then heated to 25 °C and stirring continued for a further 10 hours. The reaction mixture was slowly poured into crushed ice with stirring for a few moments. It was then diluted with diethyl ether and washed with water and brine. The organic layer was separated and dried over sodium sulfate,and the solvent was evaporated under reduced pressure to afforded 3.0 g of hexahydroxytriphenylene 8 as a grey solid in 76percent yield, m.p. >302°C. 13C NMR (DMSO-d6, 100MHz) deltaC: 107.8, 121.9, 145.2. 1H NMR (DMSO-d6, 400MHz) deltaH: 7.60 (s, 6H), 9.28 (s, 6H). Anal. Calcd. forC18H12O6 (324.28): C, 66.67; H, 3.73. Found: C, 66.66; H,3.70.
With water; hydrogen iodide; In acetic acid; for 2h;Reflux;
Subsequently, to the obtained 2,3,6,7,10,11-hexamethoxytriphenylene (28.2 g, 0.069 moles), 57percent hydroiodic acid (235.3 g, 1.05 moles) and acetic acid (145 mL) were added, and the solution was refluxed for 2 hours. After completion of the reaction, the solution was cooled down to room temperature, and the precipitated crystal was collected by filtration. The crystal collected by filtration was dried under reduced pressure to give gray type B crystal of 2,3,6,7,10,11-hexahydroxytriphenylene monohydrate (20.2 g, yield: 85.5percent) (the method of JP-A-8-119894).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping