* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With ammonium hydroxide; iodine; potassium iodide; In water; at 20℃; for 16h;Inert atmosphere;
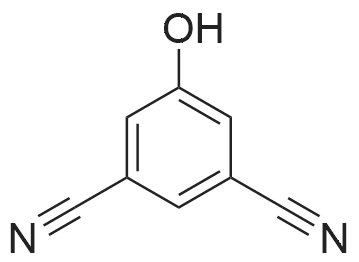
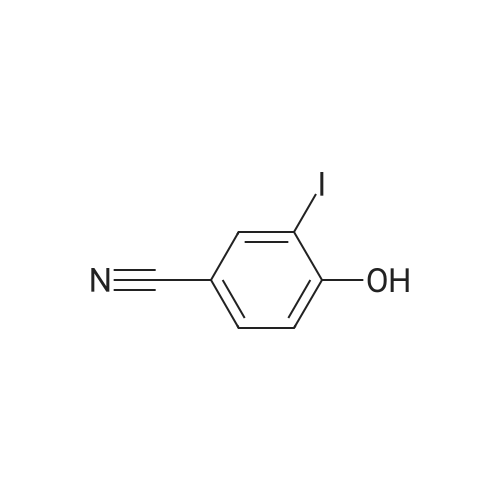
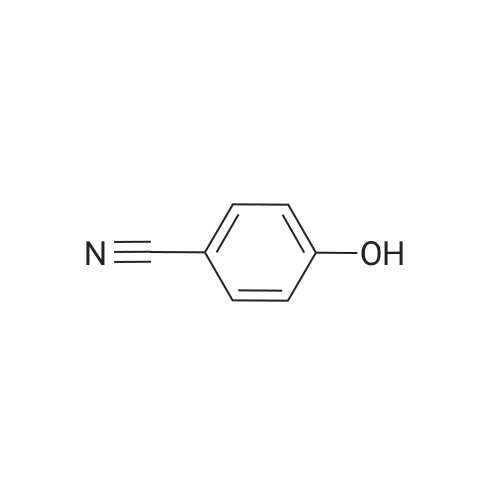
To a solution of 4-hydroxy-benzonitrile (5 g, CAS: 767-00-0) in NH4OH (225 mL) was added a solution of KI (34.14 g, CAS: 7681-11-0) and ?2(10.65 g, CAS: 7553-56-2) in H20 (50 mL). The reaction mixture was stirred at rt for 16 hours. The reaction mixture was filtered andthe filtrate was evaporated. The residue was dissolved in DCM (250 mL) and was washed with H20 (2x150 mL), saturated aqueous Na5203 solution (100 mL) and brine (100 mL). The organic layer was dried over anhydrous Na2504, filtered and concentrated under reduced pressure to give the title compound (8.44 g, 82%) that was used in the next step without further purification. LC-MS: (ESI): mlz = 244.0 [M-H]
80%
With ammonium hydroxide; iodine; potassium iodide; In water; for 6h;
To a solution of A- hydroxybenzonitrile (0.5 g; 4.18 mmol) in 25% NH4OH (22 ml) a solution of I2 (1.06 g; 4.18 mmol) and Kl (3.41 g; 20.54 mmol) in H2O (5 ml) was added at once with stirring. The stirring was continued for 6 h, during which time the mixture turn from black into colourless. The precipitate formed was filtered off and filtrate was evaporated to dryness under reduced pressure. The residue was treated with H2O (3 ml). The precipitate formed was filtered off, washed with cold H2O (3 x 2 ml), and dried in vacuo to give the title compound (0.82 g; 80%), as colourless solid. 1H-NMR (CDCI3) 7.96 (d, 1 H, 1 .9 Hz); 7. 53 (dd, 1 H, J = 1 .9 Hz, 8.5 Hz); 7.03 (d, 1 H, J = 8.5 Hz); 6.03 (s, 1 H);
80%
With ammonia; iodine; potassium iodide; In water; for 6h;
To a solution of 4- hydroxybenzonitrile (0.5 g; 4.18 mmol) in 25% NH4OH (22 ml) a solution of I2 (1.06 g; 4.18 mmol) and Kl (3.41 g; 20.54 mmol) in H2O (5 ml) was added at once with stirring. The stirring was continued for 6 h, during which time the mixture turn from black into colourless. The precipitate formed was filtered off and filtrate was evaporated to dryness under reduced pressure. The residue was treated with H2O (3 ml). The precipitate formed was filtered off, washed with cold H2O (3 x 2 ml), and dried in vacuo to give the title compound (0.82 g; 80%), as colourless solid. 1 H-NMR (CDCI3 ) 6.03 (s, 1 H); 7.03 (d, 1 H, J = 8.5 Hz); 7. 53 (dd, 1 H, J = 1 .9 Hz, 8.5 Hz); 7.96 (d, 1 H, 1 .9 Hz).
80%
With ammonium hydroxide; iodine; potassium iodide; In water; for 6h;
To a solution of 4- hydroxybenzonitrile (0.5 g; 4.18 mmol) in 25% NH4OH (22 ml) a solution of l2 (1 .06 g; 4.18 mmol) and Kl (3.41 g; 20.54 mmol) in H20 (5 ml) was added at once with stirring. The stirring was continued for 6 h, during which time the mixture turn from black into colourless. The precipitate formed was filtered off and filtrate was evaporated to dryness under reduced pressure. The residue was treated with H20 (3 ml). The precipitate formed was filtered off, washed with cold H20 (3 x 2 ml), and dried in vacuo to give the title compound (0.82 g; 80%), as colourless solid. 1 H-NMR (CDCI3) 7.96 (d, 1 H, 1 .9 Hz) ; 7. 53 (dd, 1 H, J = 1 .9 Hz, 8.5 Hz) ; 7.03 (d, 1 H, J = 8.5 Hz); 6.03 (s, 1 H) ;
With ammonia; iodine; In methanol; water; at 20℃; for 2h;
A. 3-Iodo-4-hydroxybenzonitrile 11.9 g (0.1 mol) of 4-hydroxybenzonitrile are dissolved in 250 ml of methanol and 250 ml of 20% aqueous ammonia are added. A solution of 31.75 g of iodine in 250 ml of methanol is then added dropwise, with care, due to the explosive nature of the reaction. After addition, the mixture is stirred for 2 hours at ambient temperature. The methanol is evaporated off, dilution is carried out in water and acidification with a hydrochloric acid solution is performed until pH=2 to 3 is obtained. Extraction with ethyl acetate and washing with water, a sodium thiosulfate solution and a saturated sodium chloride solution are subsequently carried out. In this way, 24.73 g of desired compound are obtained. M.p.: 144-146 C. The compound below was prepared using the same process as above:
With copper(l) iodide; potassium carbonate; dimethylbiguanide; In acetonitrile; at 20 - 60℃; for 24.1667h;
General procedure: To a 25 mL flask containing a mixture of CuI (19.2 mg,0.1 mmol), metformin (0.1 mmol), phenol (1.0 mmol), and K2CO3(2 mmol, 276 mg) in CH3CN (5 mL) was added an aryl halide(1.1 mmol). The mixture was stirred for 10 min at room temperature, and then heated to 60°C for the appropriate amount of time(see Table 4). The progress of the reaction was monitored by TLC. After completion of the reaction, the mixture was extracted with EtOAc (5 1 mL) and the organic phase separated and evaporated. Further purification by column chromatography gave the desired coupled product.
7%
With tetrabutylammomium bromide; caesium carbonate; In dimethyl sulfoxide; at 120℃; for 20h;
General procedure: Polymer supported Cu(II) catalyst (0.05 g, 0.0098 mmol) in DMSO (5 mL) was taken in a 100 ml R.B flask and stirred at room temperature for 10 min. Then aryl halide (1 mmol), phenol(1 mmol), tetrabutylammonium bromide (tBu4NBr) (0.1 mmol),Cs2CO3 (1 mmol) and DMSO (5 mL) were added to it. The final reaction mixture was refluxed at 120 C under an open air condition.The reaction mixtures were collected at different time intervals and identified by GCMS and quantified by GC. After the completion of the reaction, the catalyst was filtered off and washed with water followed by acetone and dried in oven. The filtrate was extracted with ethyl acetate (3 x 20 ml) and the combined organic layers were dried with anhydrous Na2SO4 by vacuum. The filtrate was concentrated by vacuum and the resulting residue was purified by column chromatography on silica gel to provide the desired product.
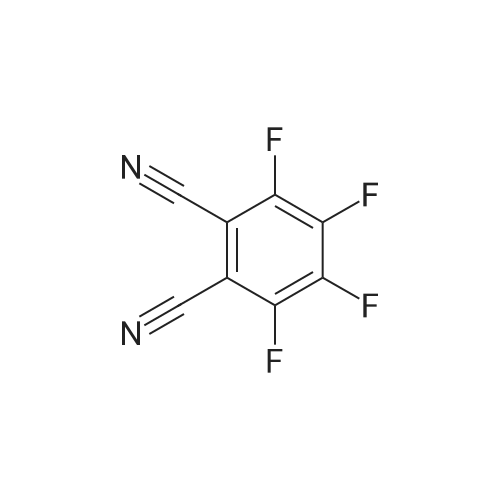
3-(2,6-dibutyl-4-methylphenoxy)-4,5-bis(4-cyanophenoxy)-6-fluorophthalonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78.3 mol%
With potassium fluoride; In acetone;
SYNTHESIS EXAMPLE 4 Synthesis of 3-(2,6-dibutyl-4-methylphenoxy)-4,5-bis(4-cyanophenoxy)-6-fluorophthalonitrile In a four-neck separable flask having an inner volume of 500 ml, 60 g (0.30 mol) of <strong>[1835-65-0]tetrafluorophthalonitrile</strong>, 41.8 g (0.72 mol) of potassium fluoride, and 160 ml of acetone. Further, to a dropping funnel attached thereto, 71.5 g (0.60 mol) of 4-cyanophenol and 110 ml of acetone were placed. The 4-cyalophenol/acetone mixed solution was added dropwise from the dropping funnel to the flask over a period of about two hours while kept stirred at -1° C. The stirring was subsequently continued for about two hours. Thereafter, the contents of the flask were stirred overnight, with the reaction temperature thereof slowly raised to room temperature. Then, to this flask, 79.8 g (0.30 mol) of 2,6-dibutyl-4-methylphenol, 20.9 g (0.36 mol) of potassium fluoride, and 15.0 mol of acetone were charged and the mixture was kept stirred at 40° C. for 10 hours. The reaction solution was cooled and filtered. The filtrate was distilled by a rotary evaporator to expel the acetone and was recrystallized from methanol. The produced crystals were separated by filtration and vacuum dried to afford 151.3 g of 3-(2,6-dibutyl-4-methylphenoxy)-4,5-bis(4-cyanophenoxy)-6-fluorophthalonitrile (yield: 78.3 mol percent).
3-(2,6-dimethylphenoxy)-4,5-bis(4-cyanophenoxy)-6-fluorophthalonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77.5 mol%
With potassium fluoride; In acetone;
SYNTHESIS EXAMPLE 5 Synthesis of 3-(2,6-dimethylphenoxy)-4,5-bis(4-cyanophenoxy)-6-fluorophthalonitrile In a four-neck separable flask having an inner volume of 500 ml, 60 g (0.30 mol) of <strong>[1835-65-0]tetrafluorophthalonitrile</strong>, 41.8 g (0.72 mol) of potassium fluoride, and 160 ml of acetone. Further, in a dropping funnel attached thereto, 71.5 g (0.60 mol) of 4-cyanophenol and 110 ml of acetone were placed. The 4-cyalophenol/acetone mixed solution was added dropwise from the dropping funnel to the flask over a period of about two hours while kept stirred at -1° C. The stirring was subsequently continued for about two hours. Thereafter, the contents of the flask were stirred overnight, with the reaction temperature thereof slowly raised to room temperature. Then, to this flask, 36.7 g (0.30 mol) of 2,6-dimethylphenol, 20.9 g (0.36 mol) of potassium fluoride, and 15.0 ml of acetone were charged and the mixture was kept stirred at 40° C. for 10 hours. The reaction solution was cooled and filtered. The filtrate was distilled by a rotary evaporator to expel the acetone and was recrystallized from methanol. The produced crystals were separated by filtration and vacuum dried to afford 116.3 g of 3-(2,6-dimethylphenoxy)-4,5-bis(4-cyanophenoxy)-6-fluorophthalonitrile (yield: 77.5 mol percent).
With potassium tert-butylate; In dimethyl sulfoxide; at 40 - 45℃;
General procedure: Phenol (94mg, 1mmol) and bromobenzene (314mg, 2.0mmol) were added to a single necked flask containing DMSO (1mL) and resulted reaction mixture was stirred for 5min. After this potassium tert-butoxide (280mg, 2.5mmol) was added portion wise and stirring continued for 6-8h at 40-45C. The progress of reaction was monitored by TLC. Upon completion of the reaction, mixture poured into water, and extracted four times with 20mL of ethyl acetate. The combined organic layer was dried over Na2SO4, and filtered. The solvent was removed in vacuo and the residue was purified by column chromatography (silica gel, eluent: hexane/ethyl acetate) to afford the coupling product.
19%
With potassium phosphate; 2-((o-toluidino)methyl)phenol; copper(l) chloride; In acetonitrile; at 81℃; for 24h;Schlenk technique; Inert atmosphere;Catalytic behavior;
General procedure: Aminophenol (5 mol %), CuCl (5 mol %), phenol (1.5 mmol), aryl halide (if solid, 1 mmol), and K3PO4 (425 mg, 2.0 mmol) were added to a screwcapped Schlenk tube under argon. The tube was then evacuated and backfilled with argon (three cycles). Aryl halide (if liquid, 1.0 mmol) and dry CH3CN (0.5 mL) were added by syringe at room temperature. The reaction mixture was stirred at needed temperature (110 oC) for 24 h. The reaction mixture was allowed to reach room temperature and then diluted with dichloromethane (10 mL). The slurry was filtered, and filter cake was washed with 10 mL of dichloromethane. The solvent was removed in vacuo, and the residue was purified by column chromatography on silica gel to afford the desired product.
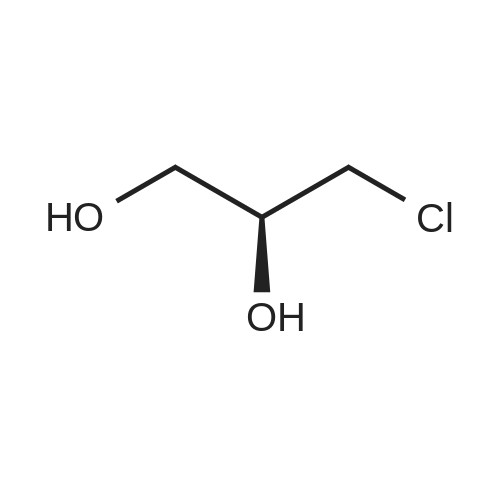
With potassium carbonate; In ethanol; water; at 80℃; for 16h;
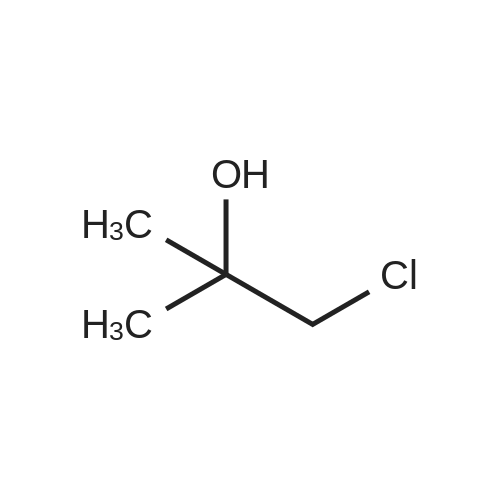
A mixture of 1-chloro-2-methyl-propan-2-ol (10 mL ), 4-hydroxybenzonitrile (2 g, 16.8 mmol), K2CO3 (9.3 g, 67.3 mmol) in water (6 mL) and ethanol (60 mL) was heated at 80 °C for 16 hours. The reaction mixture was cooled and the solvent was concentrated in vacuo. The residue was diluted with ether (200 mL) and filtered and the filtrate was washed sequentially with water (50 mL) and brine solution (50 mL). The organics were separated and dried over MgSO4 and solvent was removed in vacuo to give a residue which was purified by silica gel column chromatography using (0-100percent) EtOAc/DCM as eluent to give 4-(2-hydroxy- 2-methyl-propoxy)benzonitrile (3.0 g, 94 percent) as a yellow solid. ESI-MS m/z calc. 191.1, found 192.3 (M+1)+; Retention time: 1.05 minutes (3 min run).
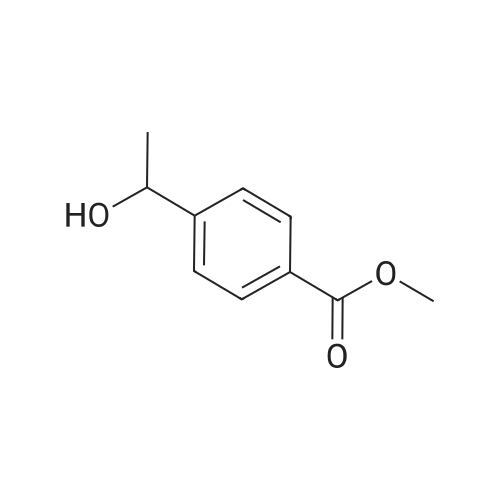
methyl 4-(1-(4-cyanophenoxy)ethyl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
45%
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 20℃; for 12h;
Methyl 4-(l-(4-cyanophenoxy)ethyl)benzoate. Methyl 4-(l -hydroxy ethyl)benzoate (540 mg, 3 mmol), 3-hydroxybenzonitrile (360 mg, 3 mmol), triphenylphosphine (1.04 mg, 4 mmol), azodicarboxylic acid diisopropyl ester (800 mg, 4 mmol) in tetrahydrofuran (130 mL) were stirred at 20C for 12 hours. Water was added and the mixture was extracted with ethyl acetate (150 mL x 3), the combined organic phase was dried by sodium sulfate, then filtered. The filtrate was evaporated in vacuum and the residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate = 3: 1) to give methyl 4-(l-(4- cyanophenoxy)ethyl)benzoate (370 mg, 45%) as colorless oil. LRMS (M + H+) m/z: calcd 281.11; found 281.).
With potassium carbonate; In N,N-dimethyl-formamide; at 130℃; under 2400.24 Torr; for 0.5h;Microwave irradiation;
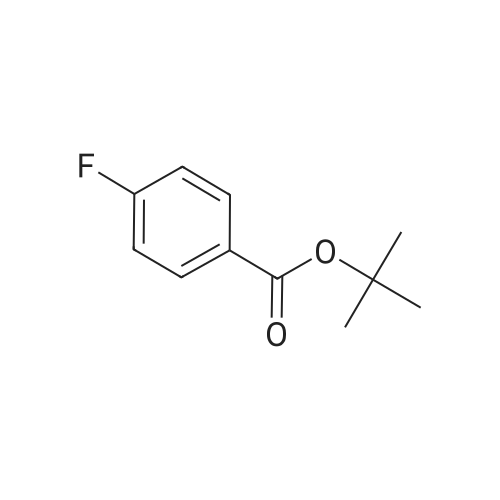
Tert-butyl 4-(4-cyanophenoxy)benzoate. To a solution of tert-butyl 4- fluorobenzoate (392 mg, 2 mmol) in dimethyl formamide (20 mL) was added 4- hydroxybenzonitrile (240 mg, 2 mmol) and potassium carbonate (552 mg, 4 mmol), the mixture was heated to 130C for 0.5 hour by microwave (pressure: 3.2 bar, equipment power : 150W). The solvent was evaporated in vacuo and purified by column chromatography (silica gel, dichloromethane/methanol = 20: 1) to give tert-butyl 4-(4-cyanophenoxy)benzoate (80 mg, 14%). LRMS (M + H+) m/z: calcd 295.12; found 295.
14%
With potassium carbonate; In N,N-dimethyl-formamide; at 130℃; under 2400.24 Torr; for 0.5h;Microwave irradiation;
To a solution of <strong>[58656-98-7]ter<strong>[58656-98-7]t-butyl 4-fluorobenzoate</strong></strong> (392 mg, 2 mmol) in dimethyl formamide (20 mL) was added 4-hydroxybenzonitrile (240 mg, 2 mmol) and potassium carbonate (552 mg, 4 mmol), the mixture was heated to 130 C. for 0.5 hour by microwave (pressure: 3.2 bar, equipment power: 150 W). The solvent was evaporated in vacuo and purified by column chromatography (silica gel, dichloromethane/methanol=20:1) to give tert-butyl 4-(4-cyanophenoxy)benzoate (80 mg, 14%). LRMS (M+H+) m/z: calcd 295.12. found 295.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping