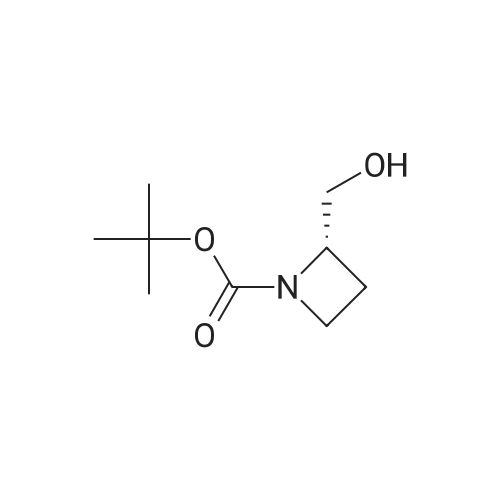
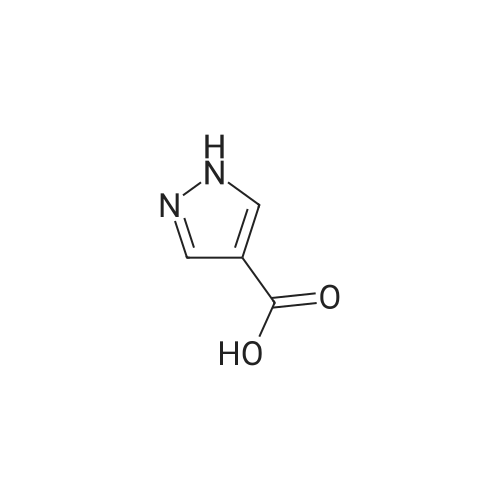
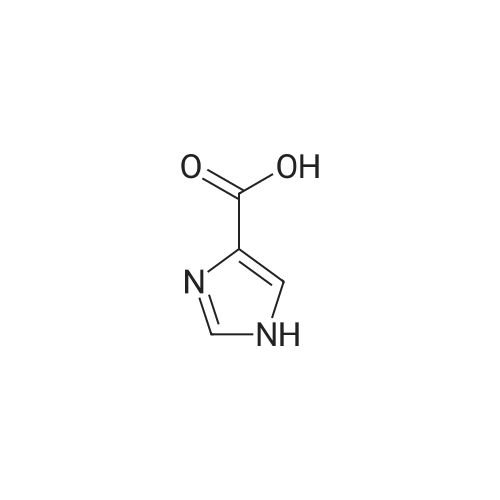
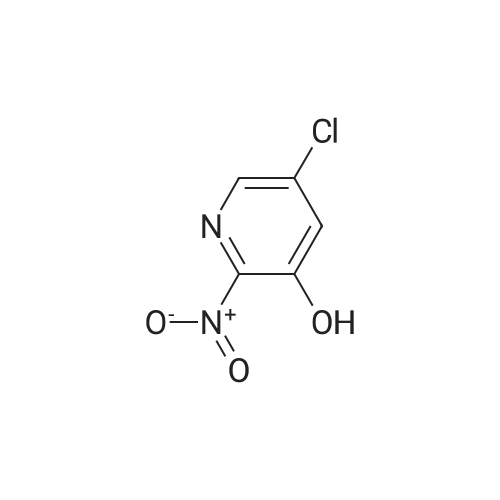
| 82% |
With sulfuric acid; nitric acid; at 0 - 20℃; |
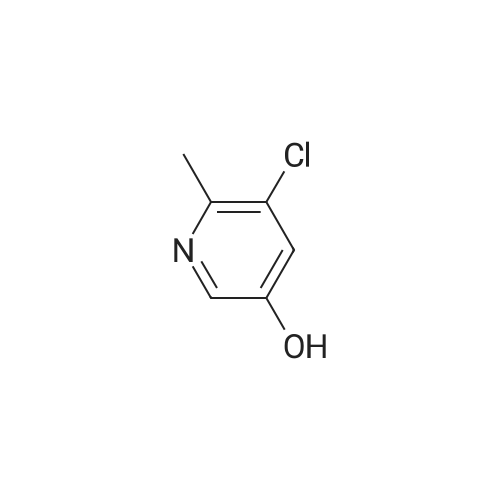
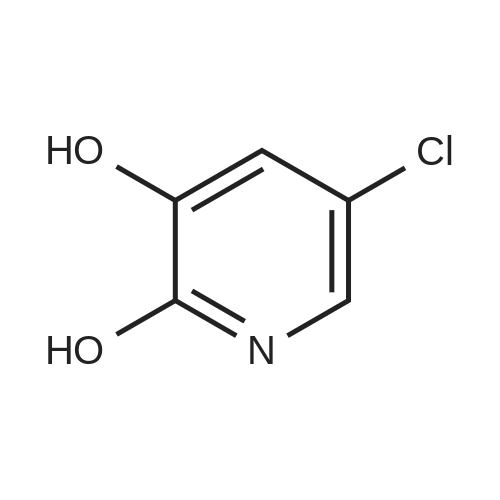
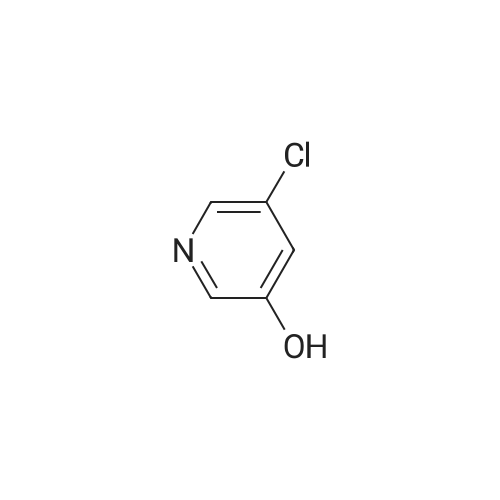
With ice cooling, 30 g of 5-chloropyridin-3-ol (232 mmol, 1 equivalent) were dissolved in 228 ml of concentrated sulphuric acid, and 24 ml of concentrated nitric acid were added slowly at 0 C. The mixture was warmed to RT, stirred overnight and then stirred into an ice/water mixture and stirred for another 30 min. The solid was filtered off, washed with cold water and air-dried. This gave 33 g (82% of theory) of the title compound which was used without further purification for the next reaction. LC-MS (Method 1): Rt=0.60 min MS (ESneg): m/z=172.9/174.9 (M-H)- 1H-NMR (400 MHz, DMSO-d6): delta=7.71 (d, 1H), 8.10 (d, 1H), 12.14 (br. 1H). |
| 82% |
With sulfuric acid; nitric acid; at 0 - 20℃; |
With ice cooling, 30 g of 5-chloropyridin-3-ol (232 mmol, 1 equivalent) were dissolved in 228 ml of concentrated sulphuric acid, and 24 ml of concentrated nitric acid were added slowly at 00 C. The reaction was warmed to RT, stirred overnight and then stirred into an ice/water mixture and stirred for another 30 mm. The solid was filtered off, washed with cold water and air-dried. This gave 33 g (82% of theory) of the title compound which was used without further purification for the next reaction.10524] LC-MS (Method 1): R=0.60 mm10525] MS (ESneg): mlz=172.9/174.9 (M-H)10526] ?H-NMR (400 MHz, DMSO-d5): oe=7.71 (d, 1H);8.10 (d, 1H); 12.14 (bt 1H). |
| 82% |
With sulfuric acid; nitric acid; at 0 - 20℃; |
With ice cooling, 30 g of 5-chloropyridin-3-ol (232 mmol, 1 equivalent) were dissolved in 228 ml of concentrated sulphuric acid, and 24 ml of concentrated nitric acid were added slowly at 0 C. The reaction was warmed to RT, stirred overnight and then stirred into an ice/water mixture and stirred for another 30 min. The solid was filtered off, washed with cold water and air-dried. This gave 33 g (82% of theory) of the title compound which was used without further purification for the next reaction. LC-MS (Method 2): Rt=0.60 min MS (ESneg): m/z=172.9/174.9 (M-H)- 1H-NMR (400 MHz, DMSO-d6): delta=7.71 (d, 1H); 8.10 (d, 1H); 12.14 (br. 1H). |
| 82% |
With sulfuric acid; nitric acid; at 0 - 20℃; |
With ice cooling, 30 g of 5-chloropyridin-3-ol (232 mmol, 1 equivalent) were dissolved in 228 ml of concentrated sulfuric acid, and 24 ml of concentrated nitric acid were added slowly at 0 C. The reaction was warmed to RT and stirred overnight. The mixture was stirred into an ice/water mixture and stirred for 30 min. The solid was filtered off, washed with cold water and air-dried. This gave 33 g (82% of theory) of the title compound which was used without further purification for the next reaction. LC-MS (Method 1): Rt=0.60 min MS (ESneg): m/z=172.9/174.9 (M-H)- 1H-NMR (400 Mhz, DMSO-d6): delta=7.71 (d, 1H); 8.10 (d, 1H); 12.14 (br. 1H). |
| 82% |
With sulfuric acid; nitric acid; at 0 - 20℃; |
With ice cooling, 30 g of 5-chloropyridin-3-ol (232mmol, 1 equivalent) were dissolved in 228 ml of concentrated sulphuric acid, and 24 ml of concentrated nitric acid were added slowly at 0 C. The reaction was warmed to RT and stirred overnight. The mixture was stirred into an ice/water mixture and stirred for 30 mm. The crystals were filtered off, washed with cold water and air-dried. This gave 33 g (82% of theory) of the title compound which was used without thrther purification for the next reaction.10372] LC-MS (Method 2): R=0.60 mm10373] MS (ESneg): mlz=172.9/174.9 (M-H)j0374] ?H-NMR (400 MHz, DMSO-d5): oe=7.71 (d, 1H); 8.10 (d, 1H); 12.14 (bt 1H). |
| 74% |
With sulfuric acid; nitric acid; In water; at 5 - 20℃; for 3 - 144h;Product distribution / selectivity; |
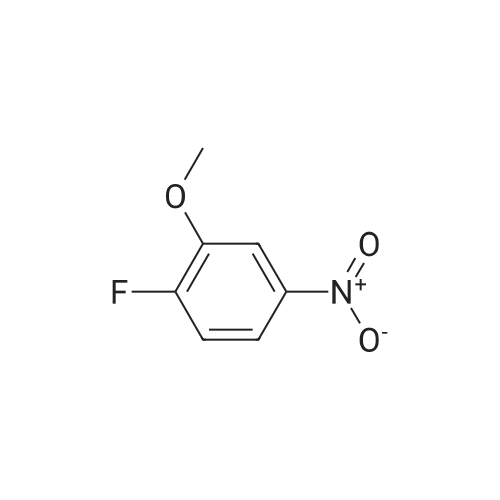
5-Chloro-3-pyridinol (15.3 mmol) is dissolved in concentrated H2SO4 (15 ml). At 5 0C concentrated nitric acid (0.9 ml) is added. The reaction mixture is allowed to warm to room temperature over 6 days. The reaction solution is pored onto ice (50 ml) and diluted with water (200 ml). The precipitate is filtered, washed with water and dried at 40 0C in vacuo. 5-Chloro-2-nitro-pyridine-3-ol is obtained as yellow powder; mp. 97; LC- MS (method B): 1.35 min, 175.1 (M+H+).; 5-Chloro-3-pyridinol (382 mmol) is dissolved in concentrated H2SO4 (375 ml). At 5 0C concentrated nitric acid (25 ml) is added. The reaction is allowed to warm to room temperature over 3 hours. The reaction solution is pored onto ice water (5000 ml). The precipitate is filtered, washed with water and dried over night at 40 0C in vacuo. 5- Chloro-2-nitro-pyridine-3-ol is obtained as yellow powder in a yield of 74 %. Mp.: 97 0C; LC-MS (Method B): 1.35 min, 175.1 (MH+). |
| 67% |
With sulfuric acid; nitric acid; at 5 - 20℃; for 3h; |
5-chloropyridin-3-ol(2.00 g, 15.4 mmol) was dissolved in concentrated H2SO4(15 mL) at 5 C. Concentrated nitric acid (1.0 mL) was then added. The reactionwas allowed to warm to room temperature over 3 hrs. The reaction solution waspoured onto ice water (25 mL). The resultant precipitate was filtered, washedwith water and dried overnight at 40 C in vacuo to afford was obtained as ayellow powder, (1.80g, 67%); H NMR (400 MHz, CDCI3) delta7.68 (d, 1H), 8.15 (d, 1H), 10.29 (s, 1H). |
|
With sulfuric acid; nitric acid; at 0 - 20℃;Cooling with ice; |
Example 27A 5-Chloro-2-nitropyridin-3-ol With ice cooling, 30 g of 5-chloropyridin-3-ol (232 mmol, I equivalent) were dissolved in 228 ml of concentrated sulphuric acid, and 24 ml of concentrated nitric acid were added slowly at 0 C. The reaction was warmed to RT and stirred overnight. The mixture was stirred into an ice/water mixture and stirred for 30 min. The solid was filtered off, washed with cold water and air-dried. This gave 33 g (82% of theory) of the title compound which was used without further purification for the next reaction. LC-MS (Method D): Rt=0.60 min MS (ESneg): m/z=172.9/174.9 (M-H)- 1H-NMR (400 MHz, DMSO-d6): delta=7.71 (d, 1H); 8.10 (d, 1H); 12.14 (br. 1H). |
| 33 g |
With sulfuric acid; nitric acid; at 0 - 20℃; |
Example 7A 5-Chloro-2-nitropyridin-3-ol With ice cooling, 30 g of 5-chloropyridin-3-ol (232 mmol, 1 equivalent) were dissolved in 228 ml of concentrated sulphuric acid, and 24 ml of concentrated nitric acid were added slowly at 0 C. The reaction was warmed to RT, stirred overnight and then stirred into an ice/water mixture and stirred for another 30 min. The solid was filtered off, washed with cold water and air-dried. This gave 33 g (82% of theory) of the title compound which was used without further purification for the next reaction. LC-MS (Method 1): Rt=0.60 min MS (ESneg): m/z=172.9/174.9 (M-H)- 1H-NMR (400 MHz, DMSO-d6): delta=7.71 (d, 1H); 8.10 (d, 1H); 12.14 (br. 1H). |
|
With nitric acid; |
Example 7A 5-Chloro-2-nitropyridin-3-ol With ice cooling, 30 g of 5-chloropyridin-3-ol (232 mmol, 1 equivalent) were dissolved in 228 ml of concentrated sulphuric acid, and 24 ml of concentrated nitric acid were added slowly at 0 C. The reaction was warmed to RT, stirred overnight and then stirred into an ice/water mixture and stirred for another 30 min. The solid was filtered off, washed with cold water and air-dried. This gave 33 g (82% of theory) of the title compound which was used without further purification for the next reaction. LC-MS (Method 1): Rt=0.60 min MS (ESneg): m/z=172.9/174.9 (M-H) 1H-NMR (400 MHz, DMSO-d6): delta=7.71 (d, 1H); 8.10 (d, 1H); 12.14 (br. 1H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping


 benzenesulfonates and their hydrochloride salts as novel water soluble antimitotic prodrugs bioactivated by cytochrome P450 1A1 in brea.png)