* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
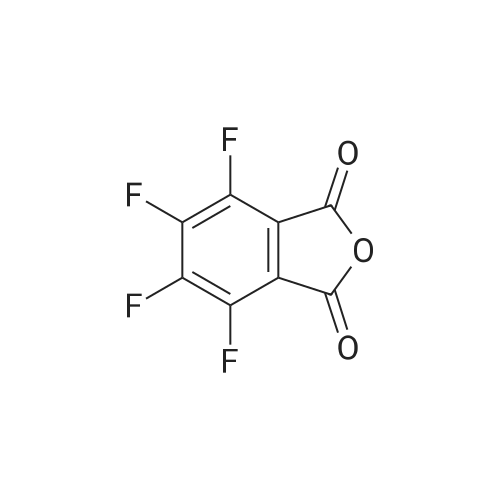
General procedure: The compounds were prepared through condensation reaction between 0.001 mol tetrafluorophthalic anhydride and 0.001 mol sulfonamides in 5 mL acetic acid under reflux at 120 C for 3-4 h. Then 20 mL distilled water was added into the reaction media. The compounds were filtered and recrystallized in ethanol.
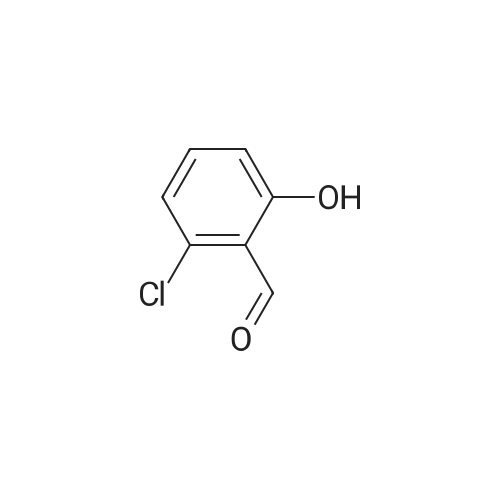
4-[(2-chloro-6-hydroxybenzylidene)amino]-N-(pyrimidin-2-yl)benzenesulfonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In methanol; at 20℃; for 5h;Reflux;
General procedure: Sulfadiazine 1 (1 mmol, 250.3 mg) was suspended in methanol (MeOH, 10 mL), and 1.1 mmol ofthe appropriate aldehyde was added in one portion under vigorous stirring. The solution was refluxedfor 5 h and then stirred at room temperature overnight. The resulting crystals were filtered off, washedwith a small amount of MeOH and then acetonitrile, and dried. The crystals were recrystallizedfrom MeOH or tetrahydrofuran/n-hexane mixture if necessary. The identity of known compounds(i.e., 2a, 2c, 2d, and 2m) was confirmed by NMR (1H and 13C) and IR spectroscopy. All spectroscopic characteristics were in accordance with previously reported data. The purity was checked additionallyby melting points measurement and elemental analysis. The numbering of hydrogen atoms in the1H-NMR spectra is depicted in Figure 3.
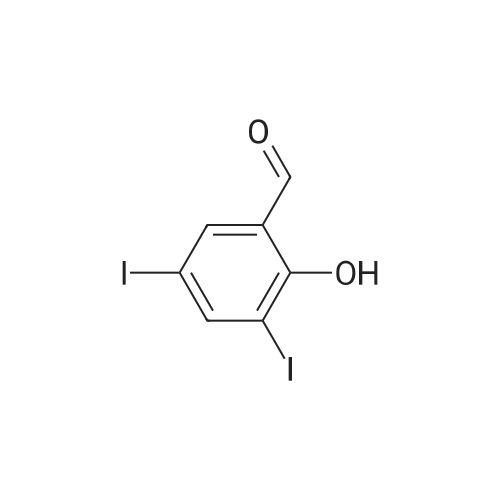
4-[(2-hydroxy-3,5-diiodobenzylidene)amino]-N-(pyrimidin-2-yl)benzenesulfonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In methanol; at 20℃; for 5h;Reflux;
General procedure: Sulfadiazine 1 (1 mmol, 250.3 mg) was suspended in methanol (MeOH, 10 mL), and 1.1 mmol ofthe appropriate aldehyde was added in one portion under vigorous stirring. The solution was refluxedfor 5 h and then stirred at room temperature overnight. The resulting crystals were filtered off, washedwith a small amount of MeOH and then acetonitrile, and dried. The crystals were recrystallizedfrom MeOH or tetrahydrofuran/n-hexane mixture if necessary. The identity of known compounds(i.e., 2a, 2c, 2d, and 2m) was confirmed by NMR (1H and 13C) and IR spectroscopy. All spectroscopic characteristics were in accordance with previously reported data. The purity was checked additionallyby melting points measurement and elemental analysis. The numbering of hydrogen atoms in the1H-NMR spectra is depicted in Figure 3.
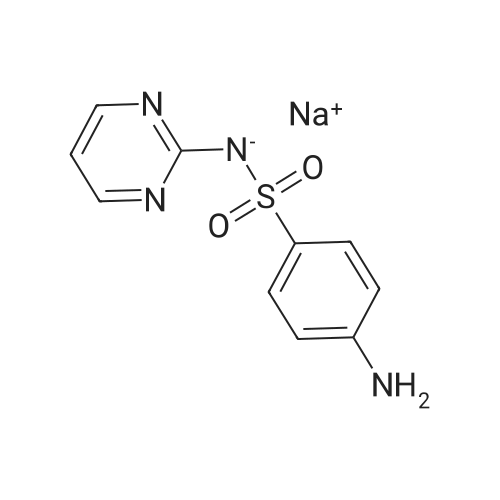
The simple salt forms were prepared by reacting 1:1 molarmixtures of sulfadiazine and MOH (M = Li, Na or K) or theorganic base in water-ethanol (50:50 v/v). The mixtures werestirred and heated to give clear solutions, before being left tocool to room temperature. Partial evaporation of these reactionmixtures over periods of 4-7 d gave suitable crystals of(I), (III), (V) and (VI), but a fine powder of Na salt (II).Good-quality crystals of (II) were obtained by vapour diffusionof ethanol into an aqueous solution of sodium sulfadiazine.Na Orange G (OG) complex (IV) was obtained bydissolving NaOG (0.20 g, 0.44 mmol) in the minimum amountof water. A slight excess of sulfadiazine (0.12 g, 0.48 mmol)was also dissolved in the minimum amount of water. The twosolutions were mixed together with stirring and acidified withconcentrated HCl. After 3 d, orange crystals of (IV) hadgrown.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping