| 91% |
|
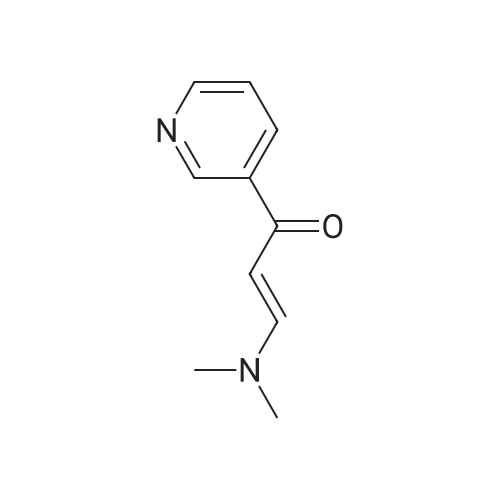
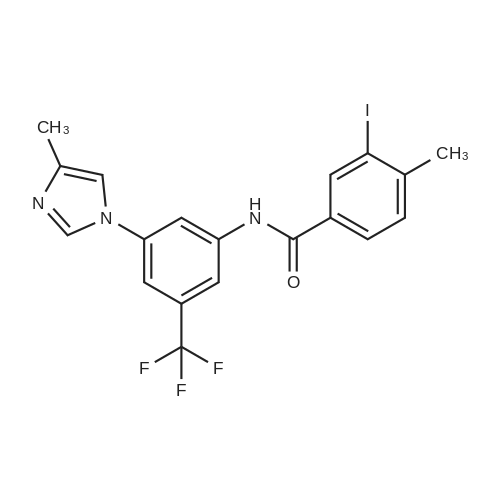
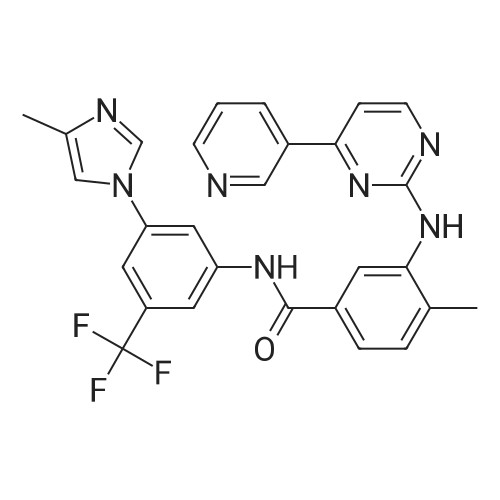
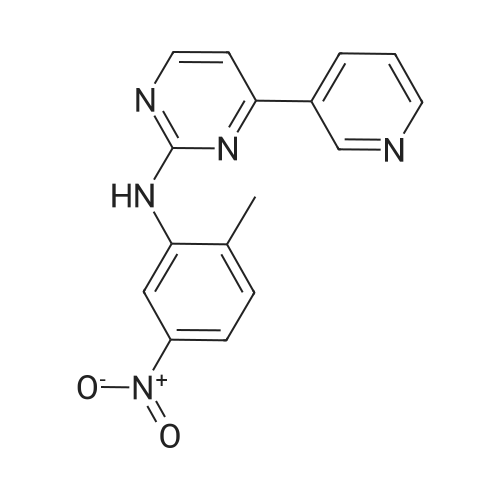
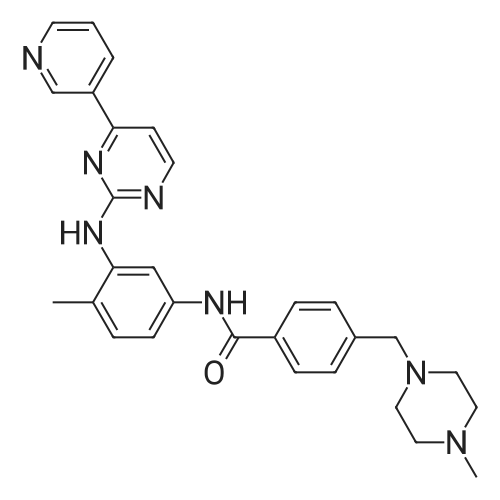
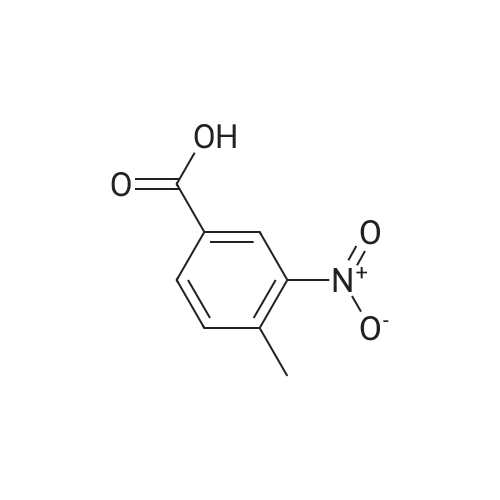
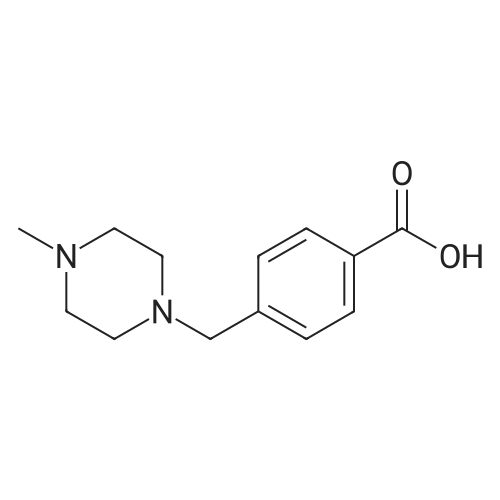
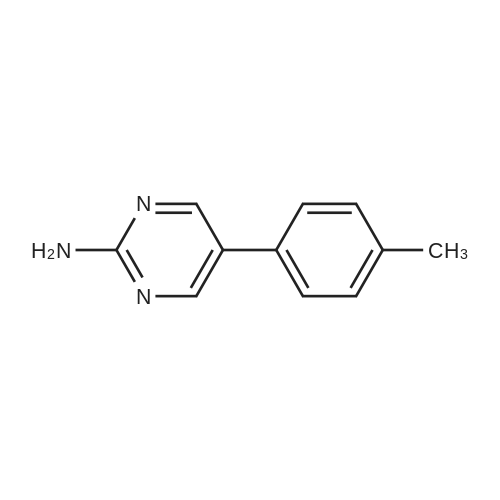
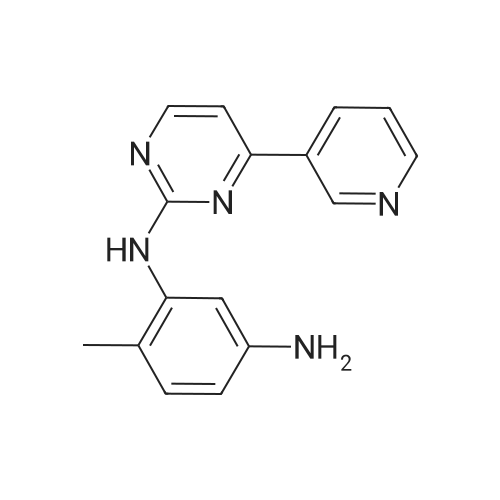
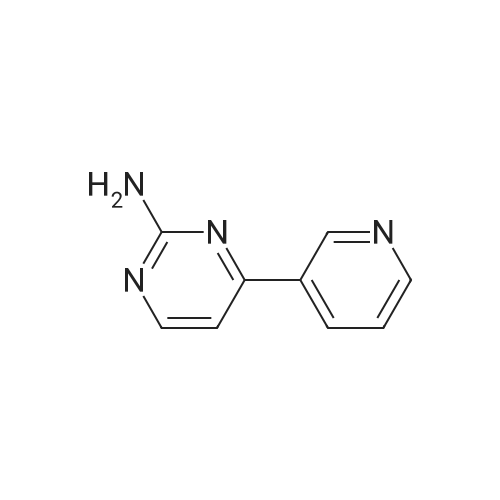
Under nitrogen protection, 40 mg of N,N'-diisopropylselenourea was uniformly mixed with 10 g of chitosan.It was placed in a tubular reactor and calcined at 500 ° C for 5 hours under nitrogen to obtain selenium and nitrogen-doped nano-carbon fibers with selenium promoting large specific surface area. The material was immersed in 0.05 mol/L copper chloride aqueous solution for 18 hours. ,Filtration, washing with water, drying to obtain a catalyst.The above catalyst is determined by ICP, wherein the mass ratio of selenium is 0.01percent, and the mass ratio of copper is 0.12percent;The specific surface area is 28 cc/g.20 mg of the above catalyst with 1.72 g of <strong>[66521-66-2]4-(3-pyridyl)-2-aminopyrimidine</strong> (see I in the following reaction formula)And 4.02g of 4-(N-methylpiperazine)methylbenzoyl (3-bromo-4-methylphenyl)amine (see II in the following reaction formula) were mixed in 20 mL of ethanol under nitrogen protection. Heat at 60 ° C for 3 hours. The catalyst is recovered by centrifugation,Concentrate the supernatant to within 5 mL, that is, a large amount of crystals are precipitated, filtered, and washed with petroleum ether.Imadinib hydrobromide was obtained in a yield of 96percent.Dissolve the salt in 20 mL of water, adjust the pH to 8.7 with 0.2 mol/L NaOH, and extract with ethyl acetate.(3 extractions per 20 mL). The organic phases were combined and dried over anhydrous sodium sulfate. After filtering, the solvent is evaporated.That is, free imatinib was obtained, and the yield was 91percent. ICP analysis indicated that the copper residue in the product was less than 0.03 ppm. |
| 72% |
With tris(dibenzylideneacetone)dipalladium(0) chloroform complex; 2,2'-bis-(diphenylphosphino)-1,1'-binaphthyl; sodium t-butanolate; In 5,5-dimethyl-1,3-cyclohexadiene; for 5.16667h;Inert atmosphere; Reflux; Sonication; |
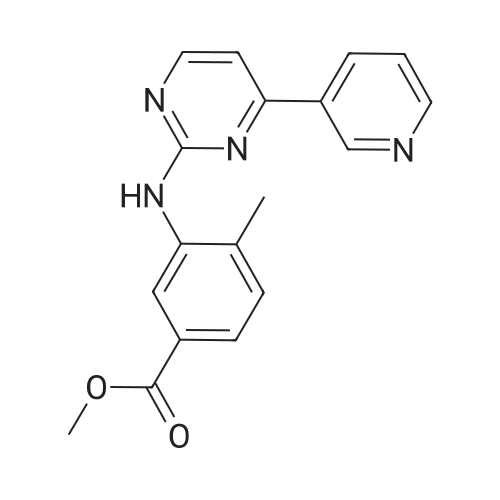
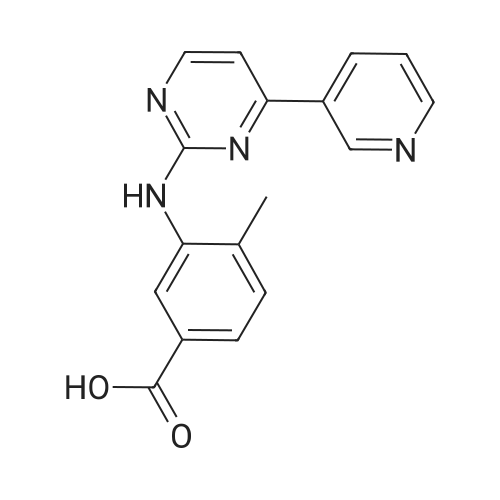
To a mixture of 4-(3-pyridyl)-2-pyrimidine-amine (172.2 mg, 1.0 mmol), N-(3-bromo-4-methyl-phenyl)-4-(4-methyl-piperazin-1-ylmethyl)-benzamide (402.4 mg, 1.0 mmol) and sodium tert.-butylate (144.2 mg, 1.5 mmol) is added a mixture of rac-BINAP (31.2 mg, 0.050 mmol) and Pd2(dba)3*CHCl3 (13 mg, 0.013 mmol) under argon. After addition of 3 ml of xylene the suspension is sonicated for 10 minutes then stirred for 5 hours under reflux. After cooling to room temperature, water (10 ml) is added to the dark brown oil and the product extracted 4 times with methylene chloride (10 ml each). The combined organic extracts are dried over MgSO4 and concentrated in vacuo. The brown oil is purified by flash-chromatography (SiO2, methanol). The product, a pale yellow solid is dissolved in methylene chloride, filtered and concentrated in vacuo. Yield: 484.3 mg of the title compound, 72percent of theory, (99.9percent area by HPLC). The product contains typically roughly 10percent of isomers which can be eliminated by preparative reversed phase chromatography. |
| 71% |
With copper(l) iodide; potassium carbonate; N,N`-dimethylethylenediamine; In 1,4-dioxane; at 100℃; for 28h;Inert atmosphere; Autoclave; |
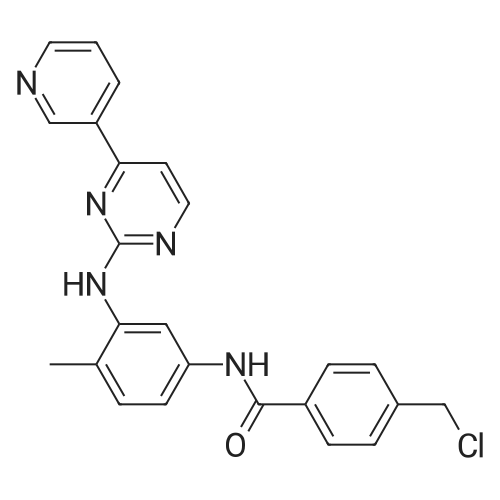
To 20 L autoclave, 12 (189.4 g, 1.1 mol), 11 (402.3 g, 1 mol), CuI(47.8 g, 0.25 mol), and K2CO3 (276.4 g, 2 mol) were added. Theautoclave was then charged with N2, and a solution of DMEDA(26.8 mL) in 12 L of 1,4-dioxane was slowly dropped. Themixture was mechanically stirred at 100 °C for 28 h. Aftercooling to room temperature, it was poured into a mixture ofconcentrated NH3 (3.5 L) and cold sat. NaCl solution (15 L, 0?5°C) and extracted by EtOAc (3 × 14 L). The combined organiclayer was dried by Na2SO4 and led to white crystal after concentration.The crystal was washed by PE and dried under vacuumovernight to afford 350.5 g of imatinib base 1 in 71percent yield. Mp 207.4?209.2 °C. IR (KBr): 3410, 3290, 2967, 2964, 2932,2801, 1628, 1588, 1532, 1507, 1478, 1450, 1416, 1380, 1346,1007, 829, 761, 700 cm?1. 1H NMR (600 MHz, DMSO-d6, TMS):delta = 10.22 (s, 1 H), 9.30 (d, J = 1.8 Hz, 1 H), 9.02 (s, 1 H), 8.71?8.70(m, 1 H), 8.54?8.50 (m, 2 H), 8.12 (s, 1 H), 7.93 (d, J = 8.4 Hz, 2H), 7.55?7.44 (m, 5 H), 7.23 (d, J = 8.4 Hz, 1 H), 3.53 (s, 2 H),2.53?2.33 (br s, 8 H), 2.25 (s, 3 H), 2.16 (s, 3 H) ppm. 13C NMR(150 MHz, DMSO-d6): delta = 165.3, 161.6, 161.2, 159.4, 151.3,148.1, 142.1, 137.8, 137.2, 134.4, 133.7, 132.2, 130.0, 128.6,127.5, 123.8, 117.2, 116.8, 107.5, 61.6, 54.7, 52.5, 45.7, 17.6ppm. Known compound: CAS Reg. No. 152459-95-5.3 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping