There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
Structure of 623-00-7 * Storage: {[proInfo.prStorage]}
Abstract: The von Hippel-Lindau (VHL) protein serves as the substrate recognition subunit of the multi-subunit Cullin-2 RING E3 ubiquitin ligase (CRL2VHL), which regulates intracellular concentrations of hypoxia inducible factors (HIFs) through a ubiquitin proteasome system (UPS) cascade. Strategic recruitment of CRL2VHL by bi- or trifunctional targeted protein degraders (e.g., PROTACs?) offers the prospect of promoting aberrant polyubiquitination and ensuing proteasomal degradation of disease-related proteins. Non-peptidic, L-hydroxyproline-bearing VHL ligands such as VH032 (1) and its chiral benzylic amine analog Me-VH032 (2), are functional components of targeted protein degraders commonly employed for this purpose. Herein, we compare two approaches for the preparation of 1 and 2 primarily highlighting performance differences between Pd(OAc)2 and Pd-PEPPSI-IPr for the key C–H arylation of 4-methylthiazole. Results from this comparison prompted the development of a unified, five-step route for the preparation of either VH032 (1) or Me-VH032 (2) in multigram quantities, resulting in yields of 56% and 61% for 1 and 2, respectively. Application of N-Boc-L-4-hydroxyproline rather than N-tert-butoxycarbonyl to shield the benzylic amine during the coupling step enhances step economy. Additionally, we identified previously undisclosed minor byproducts generated during arylation steps along with observations from amine deprotection and amidation reaction steps that may prove helpful not only for the preparation of 1 and 2, but for other VHL recruiting ligands, as well.
Abstract: Heterogenous catalysts with confined nanoporous catalytic sites are shown to have high activity and size selectivity. A solution-processable nanoporous organic polymer (1-BPy-Pd) catalyst displays high catalytic performance (TON > 200K) in the heterogeneous Suzuki–Miyaura coupling (SMC) reaction and can be used for the preparation of the intermediates in the synthesis of pharmaceutical agents. In comparison to the homogeneous catalyst analogue (2,2′-BPy)PdCl2, the heterogenous system offers size-dependent catalytic activity when bulkier substrates are used. Furthermore, the catalyst can be used to create catalytic impellers that simplify its use and recovery. We found that this system also works for applications in heterogenous Heck and nitroarenes reduction reactions. The metal-binding nanoporous polymer reported here represents a versatile platform for size-selective heterogeneous and recyclable catalysts.
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sodium azide; ammonium cerium (IV) nitrate In N,N-dimethyl-formamide at 110℃; for 6 h; Inert atmosphere; Green chemistry
General procedure: sodiumazide (1.5 mmol) was added to a magnetically stirred solution of nitrile 1a(1 mmol) in anhydrous DMF and the CAN (10 mmol percent) was added. The reactionmixture was constantly stirred for another 6 h at 110 C under nitrogenatmosphere. After the completion of reaction as seen by TLC, the reactionmixture was brought to room temperature and the solvent was evaporatedunder vacuum. The crude thus obtained, was dissolved in ethyl acetate (20 mL)and solution was washed with acidified water (4 M HCl, 15 mL) twice.Separated organic layer was washed with brine solution dried overanhydrous Na2SO4, and solvent was removed under high vacuum to obtaintetrazole 1b as a white crystalline solid in 97percent yield.
98%
With bismuth(III) chloride; sodium azide In water; isopropyl alcohol at 160℃; for 4 h; Microwave irradiation
General procedure: 2-Furonitrile 1m (186 mg, 2 mmol), NaN3 (260 mg, 4 mmol), BiCl3 (126 mg, 0.4 mmol), and 8 mL of a 3:1 isopropanol/water mixture were added to a 30-mL Pyrex microwave vessel, which was then capped. The microwave vessel was then placed in a Milestone Start Synth microwave reactor. The reaction was magnetically stirred and heated for 1 h at 150°C. The reaction was monitored by thin-layer chromatography (TLC) using an ether/hexane mixture (typically 50/50) for development. The reaction mixture was then diluted with saturated aqueous sodium bicarbonate (20 mL) and was hed with ethyl acetate (2×15 mL). The aqueous sodium bicarbonate layer was cooled with ice and acidified to a pH of 2 or less with concentrated hydrochloric acid, which was added dropwise. The precipitate formed was extracted with ethyl acetate (3×15 mL). The combined organic layers were dried with anhydrous sodium sulfate and decanted into a tared round-bottom flask. The organic layer was concentrated under reduced pressure by rotary evaporation at 40°C and then under high vacuum. The tetrazole product was recrystallized from ethyl acetate and hexane.
98%
at 120℃; for 0.5 h; Green chemistry
General procedure: To a suspension of the catalyst (0.004 g) in PEG (2 mL), nitrile (1 mmol) and sodium azide (1.2 mmol) were added and the mixture was stirred vigorously at 120 °C for the required time (Table 6). After the reaction was completed (as monitored by TLC), the catalyst was separated with a magnet. HCl (4 N, 10 mL) was then added to the residue, and the tetrazole was extracted with ethyl acetate. The organic extract was washed with distilled water, dried over anhydrous Na2SO4 and then evaporated to give the desired tetrazole.
97.2%
at 85℃; for 18 h;
at 85 °C, a mixtureof Tetrabutylammonium fluoride trihydrate (TABF3H2O) (1.31 g,5 mmol), 4-bromobenzonitrile (1.82 g, 10 mmol) and trimethylsilylazide (TMSN3) (1.73 g, 15 mmol) was under vigorous stirring conditionsfor 18 h. After completion of the reaction, the mixture was allowedto cool to room temperature, added with ethyl acetate and washedby 3×150 mL hydrochloric acid. The organic layer was isolatedand the remaining aqueous phase was further extracted with ethylacetate. The organic phases were combined and dried using anhydrousMgSO4 to afford 5-(4-bromophenyl)-1H-tetrazole. Yield 97.2percent. Meltingpoint (MP): 260–261 °C. 1H NMR (400 MHz, DMSO?d6): δ8.19–7.89 (m,2H), 7.83 (d, J=8.6 Hz, 2H). 13C NMR (400 MHz, DMSO?d6): δ 132.98(s), 129.36 (s), 125.20 (s).
95%
With sodium azide; ammonium acetate In N,N-dimethyl-formamide at 70℃; for 3 h;
General procedure: The [AMWCNTs-O–Cu(II)–PhTPY] heterogeneous catalyst was subjected to 5 successive reuses under the reaction conditions: For each reaction, nitrile (1.0mmol), NaN3 (1.3mmol) and NH4OAc (1.0mmol) were mixed and stirred in DMF (1mL) in the presence of 4mol-percent of [AMWCNTs-O–Cu(II)–PhTPY] at 70°C in an uncapped vial. After the completion of the reaction, as monitored by TLC using n-hexane/ethyl acetate, the mixture was diluted by H2O (5mL), then the mixture was vacuum-filtered onto a sintered-glass funnel, and the residue was consecutively washed with ethyl acetate (30mL), water (5mL). The heterogeneous catalyst was recharged for another reaction run. The combined supernatant and organic washings were extracted with ethyl acetate (3×10mL), the combined organic layer was dried over anhydrous Na2SO4. Removal of the solvent under vacuum, followed by purification on silica gel using hexane/ethyl acetate as the eluent afforded the pure products.
95%
With sodium azide In N,N-dimethyl-formamide at 90℃; for 3.5 h;
General procedure: In a double-necked round bottom flask (100 mL) equippedwith a condenser was added a mixture consisting ofnitrile (0.005 mol), NaN3 (0.006 mol), and monodisperse Pt NPsVC in DMF (1.5 mL). The mixture washeated at reflux until TLC monitoring indicated no furtherimprovement in the conversion. The reaction mixture wasthen cooled to room temperature, vacuum-filtered usinga sintered-glass funnel and the residue was washed withethyl acetate (3×10 mL). The filtrate was treated with5 mL HCl (4 mol L?1 to reach pH= 3 and it was allowedto stir for 30 minutes. Subsequently, the organic layer wasseparated, dried over anhydrous Na2SO4 and evaporated.The crude product was purified by recrystallization and/orcolumn chromatography on silica gel eluted with propersolvents to get pure 5-Phenyl 1H-tetrazole.
95%
With sodium azide; aminosulfonic acid In N,N-dimethyl-formamide at 120℃; for 5 h;
General procedure: A mixture of 4-nitrobenzonitrile (0.296 g, 2 mmol), sodium azide (0.195 g, 3 mmol), and sulfamic acid (0.0097 g, 0.1 mmol) was stirred at 120°C in DMF (5 mL) for the appropriate time (Table 2) until TLC (4:1 n-hexane:ethyl acetate) indicated no further progress in the conversion. After completion of the reaction (as monitored by TLC), the reaction mixture was cooled to room temperature, then 20 mL diethyl ether was added to the mixture and stirred for 10 minutes. The catalyst was separated by simple gravity filtration, washed with diethyl ether (2 £ 10 mL) and dried at 40°C for 30 min. The recovered catalyst wasused for three additional cycles and gave the tetrazole in 95, 85 and 75percent (with 4-nitrobenzonitrile). The filtrate was treated with ethyl acetate (30 mL) and 6 N HCl(20 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (20 mL). The combined organic layers were dried over anhydrous sodium sulfate and evaporated under reduced pressure to give 5-(4-nitrophenyl) tetrazole (0.363 g), 95percent yield.
94%
at 140℃; for 1.33333 h; Green chemistry
General procedure: NaN3 (0.975 g, 15 mmol)was dissolved in DES (10 mL) at room temperature by stirring until a clear solution was formed. Then benzonitrile (10 mmol) was added. The reaction mixture was constantly stirred at 140 °C and monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and poured into the cold water (10 mL). The solid was obtained and filtered. The obtained solid is taken into cold water (10 mL). Then it was acidified carefully to pH 5 with 5 M HCl. The organic material was extracted thrice with ethyl acetate; the resultant organic layer was washed with distilled water, dried over anhydrous sodium sulfate, and concentrated to give the crude solid crystalline 5-substituted 1H-tetrazole. The resulting product, although evident as a single compound by TLC, was purified by simple recrystallization from aqueous ethanol giving pure 5-substituted 1H-tetrazoles.
94%
With sodium azide; C19H17N3O4(2-)*Cu(2+) In ethylene glycol at 120℃; for 3 h;
General procedure: In 25mL round-bottomed flask, sodium azide (0.076g, 1.2mmol) and polymeric copper (II) complex (0.005g) were added to a solution of benzonitrile (0.103g, 1mmol) in ethylene glycol (3mL) with stirring at room temperature. The reaction temperature was raised up to 120°C for 3h. The reaction was monitored by TLC at regular intervals. After completion of the reaction, the reaction mixture was cooled to room temperature and treated with 10mL HCl (2N) and extracted with 10mL ethyl acetate. The resulted organic layers were separated and washed with 2×10mL distilled water, dried over anhydrous sodium sulphate and evaporated under reduced pressure. The residue was then purified by column chromatography on silica gel (100–200 mesh) to afford the corresponding products.
92%
With sodium azide In N,N-dimethyl-formamide at 110℃; for 3 h;
General procedure: A mixture of nitrile (1 mmol), sodium azide (1.5 mmol), Cu complex catalyst (0.4 molpercent) and DMF (3 mL) was taken in a round-bottomed flask and stirred at 110 °C temperature. After completion of the reaction the catalyst was separated from the reaction mixture with an external magnet and reaction mixture was treated with ethyl acetate (2 × 20 mL) and 1 N HCl (20 mL). The resultant organic layer was separated and the aqueous layer was again extracted with ethyl acetate (2 × 15 mL). The combined organic layers were washed with water, concentrated, and the crude material was chromatographed on silica gel (Hexane-EtoAc, 1:1) to afford the pure product.
92%
With indium(III) chloride; sodium azide In water; isopropyl alcohol at 160℃; for 4 h; Microwave irradiation
General procedure: Synthesis of 4-acetylbenzotetrazole (2c). 4-Acetylbenzonitrile 3c (290 mg, 2 mmol), NaN3 (260 mg, 4 mmol), InCl3(89 mg, 0.4 mmol), and 8 mL of a 3:1 isopropanol/water mixture were added to a 30-mL Pyrex microwave vessel and capped. The microwave vessel was then placed in a Milestone Start Synth microwave reactor. The reaction was magnetically stirred and heated for 1 hour at 160 oC. The pressure in the vessels was not determined. The reaction was monitored by TLC using an ether/hexane mixture (typically50/50) for development. After cooling, the reaction mixture was diluted with saturated aqueous sodium bicarbonate (20mL) and washed with ethyl acetate (2 x 15 mL). The aqueous sodium bicarbonate layer was cooled to 0 oC and acidified to a pH of 2 or less with concentrated hydrochloric acid,which was added drop-wise. The precipitate formed was extracted with ethyl acetate (3 x 15 mL). The combined organic layers were dried over anhydrous sodium sulfate and decanted into a tared round bottom flask. The organic layer was concentrated under reduced pressure. The tetrazole product was recrystallized from ethyl acetate and hexane. All reagents mentioned above were used unpurified
90%
With sodium azide; copper(II) ferrite In N,N-dimethyl-formamide at 120℃; for 12 h;
General procedure: A mixture of nitrile (1 mmol), sodium azide (1.5 mmol), catalyst (40 molpercent) and DMF (3 mL) was taken in a round-bottomed flask and stirred at 120 °C temperature for 12 h. After completion of the reaction the catalyst was separated from the reaction mixture with an external magnet and reaction mixture was treated with ethyl acetate (30 mL) and 5 N HCl (20 mL). The resultant organic layer was separated and the aqueous layer was again extracted with ethyl acetate (20 mL). The combined organic layers were washed with water, concentrated, and the crude material was chromatographed on silica gel (Hexane-EtoAc, 1:1) to afford the pure product. refText
90%
With sodium azide; copper(l) chloride In N,N-dimethyl-formamide at 120℃; for 12 h;
General procedure: A mixture of nitrile (1 mmol), sodium azide (1.5 mmol) and copper(I) chloride (4 mole percent) in DMF (2 mL) was stirred at 120 °C for the appropriate time period. The progress of the reaction was monitored by TLC. After completion, the reaction mixture was cooled and treated with 5 mL of HCl (4 mol L?1) and 10 mL of ethyl acetate, successively. The ethyl acetate extract was washed with water, dried over anhydrous sodium sulphate and concentrated under reduced pressure. The product thus obtained was recrystallised from acetic acid to afford pure 5-substituted 1H-tetrazoles.
90%
at 120℃; for 3 h;
General procedure: To a stirred mixture of sodium azide (1.2 mmol) in PEG-400(2 mL), a nitrile compound (1 mmol) and NiNP-PNF (200 mL) were added and heated at 120°C under atmospheric conditions.The reaction progress was monitored by TLC. Upon reaction completion, the mixture was allowed to cool to ambient temperature and then filtered and extracted with ethyl acetate. The organic layer was washed with 1N HCl, dried with anhydrous Na2SO4, and filtered to afford pure 5-substituted tetrazoles.
89%
With sodium azide; activated Fuller’s earth In dimethyl sulfoxide at 120℃; for 2 h; Green chemistry
General procedure: To a DMSO (3 ml) solution of nitrile (1 mmol), and sodium azide (1.5 mmol), was added catalyst (10 wt percent). The reaction mixture was stirred to 120 0C in an oil bath. The reaction was monitored by TLC. After completion of the reaction, the mixture was filtered to separate the catalyst. The filtrate was quenched with water (30 ml), acidified with 5N HCl (20 ml) to precipitate the product, extracted with ethyl acetate (2 X 20 ml). The combined organic layers were washed with water, dried over sodium sulphate and evaporated under reduced pressure to give the product.
89%
at 120℃; for 9 h;
General procedure: A mixture of the required nitrile (1 mmol), sodium azide(1 mmol) and the catalyst MNP (0.05 g) was stirred at 120° C in PEG (1 mL) as solvent. After completion of the reaction, as indicated by TLC, the mixture was cooled to room temperature and diluted with 1:1 H2O:Ethyl acetate(10 mL) and then stirred at ambient temperature (10 min). The catalyst was removed by applying a magnetic field, and the decantate was treated with HCl (4 N, 10 mL). The organic layer was separated, washed with water, dried over sodium sulfate and concentrated to precipitate the crude crystalline solid. The pure tetrazoles were characterized bytheir spectroscopic data and melting points.
88%
With sodium azide In N,N-dimethyl-formamide at 120℃; for 6.5 h;
General procedure: A mixture of nitrile (1 mmol), sodium azide (1.5 mmol) and catalyst (0.02 g, contains 0.4 molpercent of Cu(II)) in DMF (3 mL) was taken in a round-bottomed flask and stirred at 120 °C. The progress of the reaction was followed by thin-layer chromatography (TLC). After completion of the reaction, the reaction mixture was cooled to room temperature and diluted with ethyl acetate (3×20 mL). The catalyst was removed by using magnetic field or filtration and then the resulting solution was washed with 1N HCl, dried over anhydrous Na2SO4 and then was evaporated. The crude products were obtained in excellent yields. All products were characterized by 1H, 13C NMR, FT-IR, and melting point which were in agreement with literature. We have reported the spectral data of some aromatic and heteroaromatic synthesized compounds
88%
at 120℃; for 15 h; Green chemistry
General procedure: In a round-bottomed flask, a mixture of nitrile (1 mmol) and sodium azide (1.2 mmol) in the presence of 40 mg of Fe3O4*SBTU*Ni(II) was stirred at 120 °C in PEG for an appropriate time (monitored by TLC). Then, the reaction mixture was cooled down to room temperature. After magnetic separation of catalyst, HCl (4 N, 10 mL) was added to the filtrate and the product extracted with ethyl acetate (2 × 10 mL). The organic layer was washed with water several times, dried with anhydrous Na2SO4 and concentrated to give the crude solid crystalline product.
87%
With sodium azide; (1,10-phenanthroline)bis(triphenylphosphine)copper(I) nitrate In water; isopropyl alcohol at 65℃; for 0.25 h; Inert atmosphere; Microwave irradiation; Green chemistry
General procedure: In a round-bottomed flask, a mixture of organic nitrile 1 (1.0 equiv) and NaN3 (1.5 equiv) was added to 5 ml solution of H2O-IPA (1:1) containing 10 molpercent [Cu(phen)(PPh3)2]NO3 as catalyst under N2 atmosphere. The reaction mixture was irradiated under microwave heating at 245 W for 15–25 min at 65°C. Reaction progress was monitored by thin-layer chromatography (TLC). After reaction completion, the mixture was filtered to remove the catalyst. The filtrate was acidified with 5 N HCl (20 ml) to neutralize the product, extracted with ethyl acetate (2 9 10 ml). The combined organic layer was dried over anhydrous MgSO4. The combined filtrate was subjected to evaporation to obtain the crude compound, which was purified over silica gel column (60–120 mesh) using 50 percent ethyl acetate in hexane as eluent to obtain corresponding 5-substituted 1H-tetrazoles 2 as product.
86%
With sodium azide In methanol; N,N-dimethyl-formamide at 20 - 100℃; for 7 h;
General procedure: A mixture of benzonitrile (1 mmol), sodium azide (2 mmol), Ln(OTf)3-SiO2 (2008 mg) and DMF/MeOH (4:1, 5 mL) in a pressure vial was initially stirred at room temperature. After 30 min, the temperature of the reaction mixture was raised to 100 °C and stirred for another 7 h. After consumption of 1a (as indicated by TLC), the catalyst was separated by filtration and the filtrate was treated with ethyl acetate (15 mL). The organic layer was washed with 4 N HCl (20 mL). The resultant organic layer was separated and the aqueous layer was extracted with ethyl acetate (15 mL). The combined organic layer was washed with water (2 × 10 mL), dried over anhydrous sodium sulfate and concentrated to afford white crystalline solid.5-Phenyl-1H-tetrazole(3a)IR (KBr, cm?1): 3331, 2907, 2850, 2611, 1607, 1485, 1433, 1050, 828, 742. 1H NMR (300 MHz, CDCl3): 8.04–8.007 (m, 1H), 7.611–7.574 (m, 2H). 13C NMR (75 MHz, CDCl3): 156.03, 131.19, 129.08, 126.805, 123.924. MS: m/z = 146 [M]+.
82%
With trimethylsilylazide; di(n-butyl)tin oxide In 1,4-dioxane at 150℃; for 0.833333 h; Microwave irradiation
General procedure: Dibutyltin oxide (0.33 mmol, 82 mg, 0.2 equiv), andtrimethylsilyl azide (3.33 mmol, 383 mg, 2 equiv) were added to a solution of 3-bromobenzonitrile (300mg, 1.67 mmol, 1 equiv) in anhydrous 1,4-dioxane (2 mL/mmol). The reaction mixture was subjectedto microwave irradiation in a tightly sealed microwave vessel for 50 min at 150 0C, then cooled to roomtemperature. The solvent was removed under reduced pressure. The residue was dissolved in diethylether (10 mL and extracted with 2 M aq. NaOH (3 x 10 mL). The aqueous layer was acidified with 4 Maq. HCl to pH 1 and extracted with ethyl acetate (4 x 10 mL). The organic extract was washed withbrine (10 mL), dried over MgSO4, and evaporated under reduced pressure to give the intermediatetetrazole(1.45 mmol, 326 mg, 86percent) as a white solid.
79%
With sodium azide; lead(II) chloride In N,N-dimethyl-formamide at 120℃; for 8 h; Inert atmosphere
General procedure: Benzonitrile (103 mg, 1 mmol) and sodium azide (97.5 mg, 1.5 mmol) were dissolved in 2 ml of dry DMF in a 25 ml round bottom flask. PbCl2 (27.8 mg, 0.1 mmol, 10 mol percent) was added to the reaction mixture and stirred at 120 °C for 8 h under nitrogen. After completion of the reaction (as monitored by TLC), the reaction mixture was cooled to room temperature and 10 ml of ice water was added followed by addition of 3 N HCl until the reaction mixture became strongly acidic (pH 2-3). The reaction mixture was extracted three times with 20 ml ethyl acetate. The organic layer was washed with brine solution and dried over anhydrous sodium sulfate, and was evaporated under reduced pressure to give a white solid product of 5-phenyl 1H-tetrazole with 81percent yield.
79%
With sodium azide In N,N-dimethyl-formamide at 120℃; for 16 h;
General procedure: In a round-bottom flask, 0.2 g benzonitrile (2 mmol) and0.4 g sodium azide (6 mmol), were added to 10 mL DMF.To this mixture, 20 mg of functionalized KIT-6 was addedand the reaction mixture was refluxed. The progress ofreaction was monitored by TLC (75:25 ethyl acetate/nhexane).After completion of the reaction, the reactionmixture was cooled and filtered. The solid materials werewashed three times with acetone and then with the water.The catalyst was collected and dried to activation for nextrun. The product was obtained by acidification of solutionwith hydrochloric acid (5 mL, 6 M). The precipitate wasfiltered and recrystallized from a water/ethanol mixture toget pure product as a white powder, yield: 88percent.
78%
With sodium azide; silver nitrate In N,N-dimethyl-formamide at 20 - 120℃; for 5 h;
General procedure: Sodiumazide (0.378 g, 0.046 mmol) was added to a solution of AgNO3 (5 mg, 10 mmol)in DMF (5 ml) and reaction mixture was stirred for 5 min, to this stirredsolution benzonitrile 1a (0.4 ml, 0.033 mmol) was added dropwise over theperiod of 1 min at room temperature and stirring continued for 10 min at thesame temperature and then heated at 120 C for 5 h. After consumption of 1a,the reaction mixture was cooled to room temperature and chilled by addingcrushed ice into the reaction mixture followed by addition of 2 N HCl tillreaction mixture reached the pH 2. The reaction mixture was then extractedwith ethyl acetate. The organic layer was dried with anhydrous Na2SO4, andconcentrated to obtain tetrazole 2a in 83percent yield as an off white solid (268 mg).
72%
With sodium azide; tetra(n-butyl)ammonium hydrogensulfate In water at 85℃; for 9 h; Green chemistry
General procedure: General Procedure for Preparation of Tetrazoles in Water(Method II). TBAHS (0.25 mmol) was added to a mixture of nitrile (1 mmol), sodium azide (1.5 mmol), and 2 mL H2O in around-bottomed flask. The reaction mixture was heated to 85 °C. After completion of the reaction (as monitored by TLC), the crude reaction mixture was transferred into a separatory funnel, to which was added 1 N HCl (15 mL) extracted by ethylacetate (EtOAc, 10 mL × 5). The combined organic layers were washed with H2O and dried over anhydrous sodium sulfate, and were evaporated under reduced pressure to give pure 5-substituted-1H-tetrazole.
70%
With sodium azide; ammonium chloride In N,N-dimethyl-formamide for 24 h; Reflux
General procedure: In a typical procedure, 5-aryl-1H-tetrazoles (1–24) were synthesized by adding aryl nitriles (1 eq.), sodium azide (1.2 eq.), and ammonium chloride (1 eq.) in solvent, the mixture was refluxed for 24 h. Progress of the reaction was monitored by thin layer chromatography. After completion of the reaction, 2.5 mL of 2M NaOH was added and the solution was stirred for half an hour. The reaction mixture was concentrated on reduced pressure, and dissolved in water. 3M HCl was added to the reaction mixture until precipitates formed. The precipitates were filtered and washed with distilled water. The yields of title compounds were found to be moderate to high.
67%
With sodium azide In N,N-dimethyl-formamide for 24 h; Sealed tube; Green chemistry
General procedure: The benzonitrile derivatives (2 mmol), sodium azide (3.2 mmol), and DMF (2 mL) were mixed in a sealed tube, then 30 mg of catalyst MSS-SO3H (or catalyst MSS-SO3Zn) was added into the tube, which was heated for 24 h under 140 C. After 24 hours’ reaction,the catalyst was separated by magnetic force, and the solution was poured into water.The liquid was acidified to pH 1, then ethyl acetate was added to extract the tetrazoles. Carefully evaporating the solvent under reduced pressure, we got the isolated tetrazoles. A sample for characterization was purified on a flash silica column.
54%
With sodium azide; scandium tris(trifluoromethanesulfonate) In water; isopropyl alcohol at 160℃; for 1 h; Microwave irradiation; Sealed tube
General procedure: Synthesis of 5-(4-chlorophenyl)-1H-tetrazole (2c) was achieved as follows: 4-chlorobenzonitrile 1c (274 mg, 2 mmol), NaN3 (260 mg, 4 mmol), Sc(OTf)3(197 mg, 0.4 mmol), and 8mL of a 3:1 isopropanol=water mixture were added to a30-mL Pyrex microwave vessel and capped. The microwave vessel was then placedin a Milestone Start Synth microwave reactor. The reaction was magnetically stirredand heated for 1 h at 160 C. The reaction was monitored by thin-layer chromatography(TLC) using an ether=hexane mixture (typically 50=50) for development.The reaction mixture was then diluted with saturated aqueous sodium bicarbonate(20 mL) and washed with ethyl acetate (215mL). The aqueous sodium bicarbonatelayer was cooled with ice and acidified to a pH of 2 or less with concentratedhydrochloric acid, which was added dropwise. The precipitate formed was extractedwith ethyl acetate (315 mL). The combined organic layers were dried with anhydroussodium sulfate and decanted into a tared round-bottom flask. The organiclayer was concentrated under reduced pressure by rotary evaporation at 40 C andthen under high vacuum. The tetrazole product was recrystallized from ethyl acetateand hexane. All reagents mentioned were not unpurified.
Reference:
[1] Journal of the Brazilian Chemical Society, 2012, vol. 23, # 12, p. 2197 - 2203
[2] Tetrahedron Letters, 2014, vol. 55, # 44, p. 6034 - 6038
[3] Synthetic Communications, 2015, vol. 45, # 8, p. 1023 - 1030
[4] Transition Metal Chemistry, 2017, vol. 42, # 8, p. 703 - 710
[5] Angewandte Chemie - International Edition, 2010, vol. 49, # 39, p. 7101 - 7105
[6] Catalysis Science and Technology, 2015, vol. 5, # 9, p. 4452 - 4457
[7] Dyes and Pigments, 2018, vol. 158, p. 20 - 27
[8] Applied Organometallic Chemistry, 2019, vol. 33, # 1,
[9] Photochemical and Photobiological Sciences, 2014, vol. 13, # 2, p. 342 - 357
[10] RSC Advances, 2015, vol. 5, # 84, p. 68558 - 68564
[11] Journal of Organometallic Chemistry, 2013, vol. 738, p. 41 - 48
[12] New Journal of Chemistry, 2013, vol. 37, # 10, p. 3261 - 3266
[13] RSC Advances, 2015, vol. 5, # 62, p. 49849 - 49860
[14] RSC Advances, 2016, vol. 6, # 39, p. 32653 - 32660
[15] Applied Organometallic Chemistry, 2016, vol. 30, # 8, p. 705 - 712
[16] Journal of Nanoscience and Nanotechnology, 2017, vol. 17, # 3, p. 1992 - 1999
[17] Organic Preparations and Procedures International, 2017, vol. 49, # 4, p. 346 - 354
[18] New Journal of Chemistry, 2018, vol. 42, # 16, p. 13754 - 13762
[19] Heterocycles, 2014, vol. 89, # 9, p. 2137 - 2150
[20] RSC Advances, 2016, vol. 6, # 79, p. 75227 - 75233
[21] Synthetic Communications, 2017, vol. 47, # 8, p. 779 - 787
[22] Journal of Organometallic Chemistry, 2018, vol. 870, p. 16 - 22
[23] Chemistry of Heterocyclic Compounds (New York, NY, United States), 1994, vol. 30, # 10, p. 1192 - 1194[24] Chimia, 1994, # 10, p. 1375 - 1377
[25] Applied Organometallic Chemistry, 2016, vol. 30, # 11, p. 897 - 904
[26] Journal of Molecular Catalysis A: Chemical, 2014, vol. 393, p. 18 - 29
[27] RSC Advances, 2015, vol. 5, # 28, p. 21651 - 21658
[28] RSC Advances, 2016, vol. 6, # 61, p. 56638 - 56646
[29] MedChemComm, 2017, vol. 8, # 10, p. 1953 - 1964
[30] Medicinal Chemistry, 2017, vol. 13, # 4, p. 359 - 364
[31] Chemistry - An Asian Journal, 2019, vol. 14, # 5, p. 612 - 620
[32] Applied Organometallic Chemistry, 2018, vol. 32, # 8,
[33] Tetrahedron Letters, 2011, vol. 52, # 28, p. 3565 - 3569
[34] Journal of Materials Chemistry, 2012, vol. 22, # 33, p. 17227 - 17235
[35] RSC Advances, 2015, vol. 5, # 16, p. 12372 - 12381
[36] Chemical Papers, 2015, vol. 69, # 9, p. 1231 - 1236
[37] RSC Advances, 2015, vol. 5, # 126, p. 104087 - 104094
[38] RSC Advances, 2016, vol. 6, # 38, p. 31850 - 31860
[39] Applied Organometallic Chemistry, 2017, vol. 31, # 9,
[40] Australian Journal of Chemistry, 2017, vol. 70, # 10, p. 1127 - 1137
[41] Chinese Chemical Letters, 2012, vol. 23, # 2, p. 161 - 164
[42] Tetrahedron Letters, 2016, vol. 57, # 51, p. 5815 - 5819
[43] Transition Metal Chemistry, 2017, vol. 42, # 2, p. 131 - 136
[44] Journal of Organometallic Chemistry, 2013, vol. 743, p. 87 - 96
[45] Journal of Sulfur Chemistry, 2018, vol. 39, # 3, p. 237 - 251
[46] Applied Organometallic Chemistry, 2018, vol. 32, # 8,
[47] RSC Advances, 2016, vol. 6, # 99, p. 96623 - 96634
[48] Research on Chemical Intermediates, 2017, vol. 43, # 12, p. 7365 - 7374
[49] Applied Organometallic Chemistry, 2017, vol. 31, # 12,
[50] RSC Advances, 2013, vol. 3, # 13, p. 4362 - 4371
[51] Tetrahedron Letters, 2014, vol. 55, # 25, p. 3557 - 3560
[52] New Journal of Chemistry, 2014, vol. 38, # 7, p. 3078 - 3083
[53] Journal of the Iranian Chemical Society, 2012, vol. 9, # 5, p. 799 - 803
[54] New Journal of Chemistry, 2015, vol. 39, # 6, p. 4814 - 4820
[55] European Journal of Medicinal Chemistry, 2018, vol. 145, p. 634 - 648
[56] E-Journal of Chemistry, 2012, vol. 9, # 3, p. 1145 - 1152
[57] Bioorganic and Medicinal Chemistry, 2017, vol. 25, # 20, p. 5278 - 5289
[58] Comptes Rendus Chimie, 2016, vol. 19, # 3, p. 305 - 312
[59] Journal of the Iranian Chemical Society, 2018, vol. 15, # 4, p. 831 - 838
[60] Tetrahedron Letters, 2014, vol. 55, # 11, p. 1879 - 1882
[61] Bulletin of the Korean Chemical Society, 2015, vol. 36, # 1, p. 198 - 202
[62] Bioorganic Chemistry, 2018, vol. 79, p. 201 - 211
[63] Synthetic Communications, 2018, vol. 48, # 20, p. 2652 - 2662
[64] Russian Journal of Organic Chemistry, 2011, vol. 47, # 5, p. 728 - 730
[65] Russian Journal of Organic Chemistry, 2007, vol. 43, # 5, p. 765 - 767
[66] Synthetic Communications, 2015, vol. 45, # 2, p. 218 - 225
[67] Patent: WO2006/38100, 2006, A1, . Location in patent: Page/Page column 80-81
[68] European Journal of Medicinal Chemistry, 2011, vol. 46, # 11, p. 5680 - 5687
[69] Chinese Chemical Letters, 2011, vol. 22, # 5, p. 599 - 602
[70] Tetrahedron Letters, 2016, vol. 57, # 5, p. 523 - 524
[71] Chemistry of Heterocyclic Compounds, 2015, vol. 51, # 11-12, p. 984 - 990[72] Khim. Geterotsikl. Soedin., 2015, vol. 51, # 11-12, p. 984 - 990,7
[73] Applied Organometallic Chemistry, 2017, vol. 31, # 5,
[74] Organic Preparations and Procedures International, 2018, vol. 50, # 5, p. 493 - 501
With pyridine; potassium dihydrogenphosphate; palladium diacetate; copper(II) bis(trifluoromethanesulfonate); 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene In N,N-dimethyl acetamide at 120℃; for 12 h;
Add N,N-dimethylacetamide (200 mL) to a 500 mL three-necked flask.2-(4-Cyanophenyl)formic acid (II) (35.1 g, 0.2 mol), 4-bromobenzonitrile (III) (72.6 g, 0.4 mol),Potassium phosphate (85.7 g, 0.4 mol), Pd(OAc) 2 (0.45 g, 0.002 mol), CuOTf (12.8 g, 0.06 mol),Pyridine (9.7 g, 0.12 mol) and 4,5-bisdiphenylphosphino-9,9-dimethyloxaxan (2.32 g, 0.004 mol),Stirring, heating to 120 ° C, reaction for 12 hours,The reaction was completely detected by HPLC (2-(4-cyanophenyl)formic acid (II) content was less than 1percent), and the temperature was lowered to room temperature.The solid was slowly added with water, and the solid was dissolved with ethyl acetate and then extracted (50 mL x 3).The resulting organic phase was washed with 5percent aqueous HCl (100 mL) and sat. sodium chloride (100 mL).Dry over anhydrous sodium sulfate, EtOAc (EtOAc m.)The product was obtained as a white solid (44.7 g, yield: 96.3percent).
Reference:
[1] Patent: CN108409604, 2018, A, . Location in patent: Paragraph 0055-0060; 0061; 0062; 0063; 0064
4
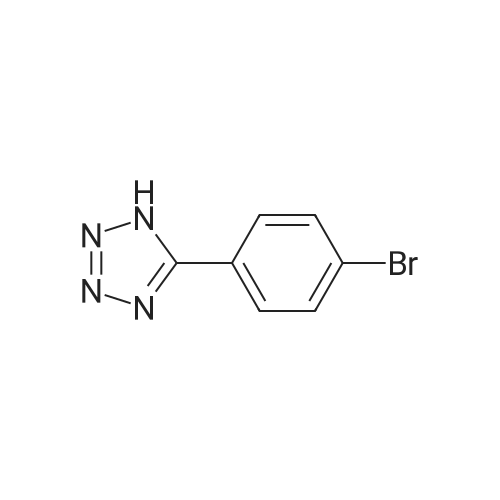
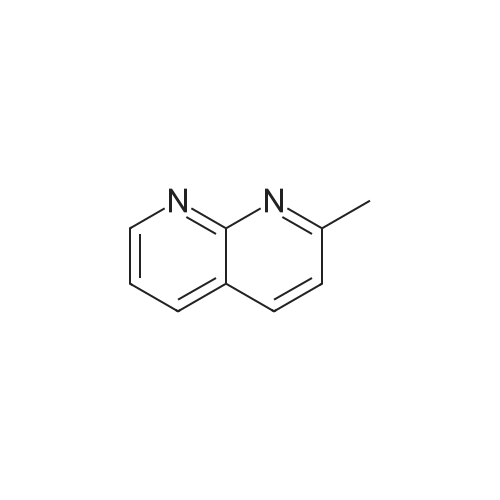
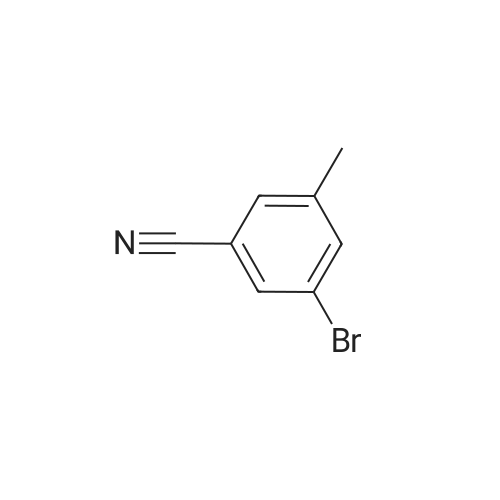
[ 623-00-7 ]
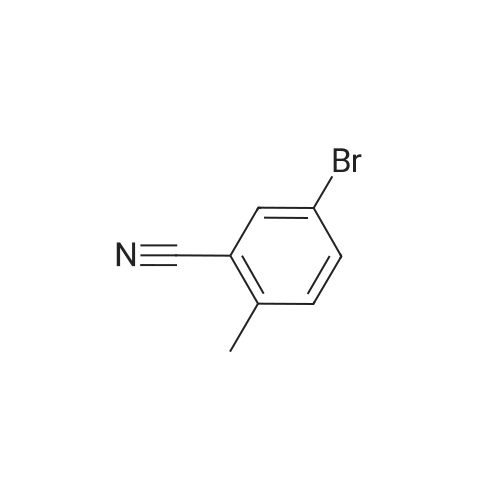
[ 108-24-7 ]
[ 1591-30-6 ]
[ 1443-80-7 ]
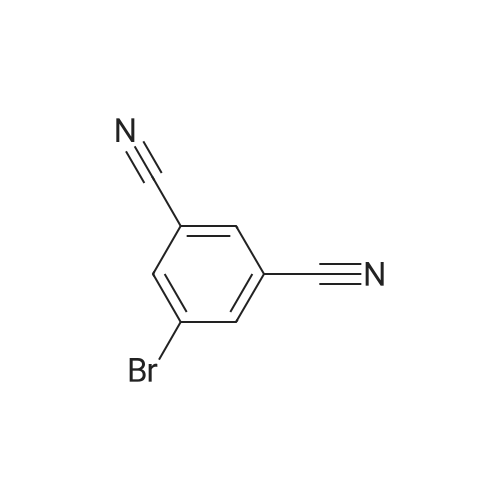
[ 32446-66-5 ]
[ 100-47-0 ]
Reference:
[1] Journal of Organic Chemistry, 2004, vol. 69, # 3, p. 936 - 942
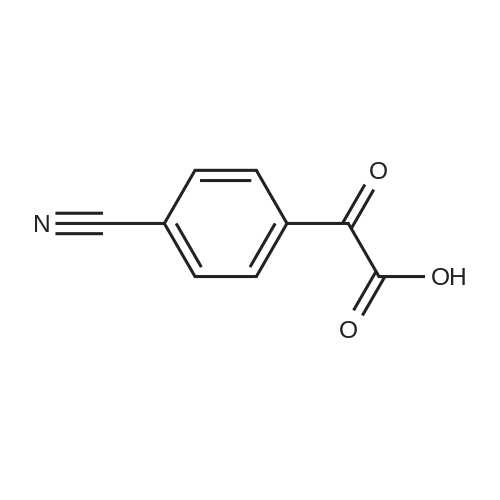
5
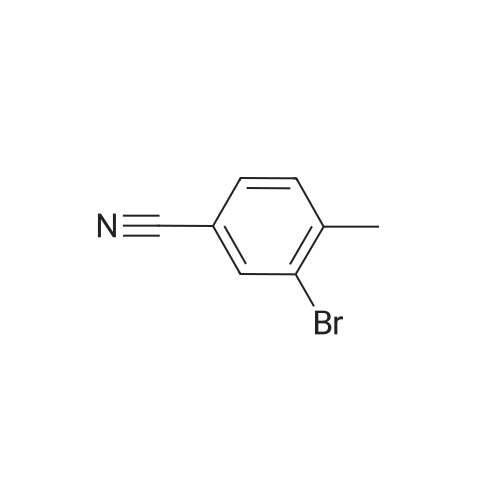
[ 623-00-7 ]
[ 98-86-2 ]
[ 32446-66-5 ]
Reference:
[1] Journal of Organic Chemistry, 2014, vol. 79, # 14, p. 6554 - 6562
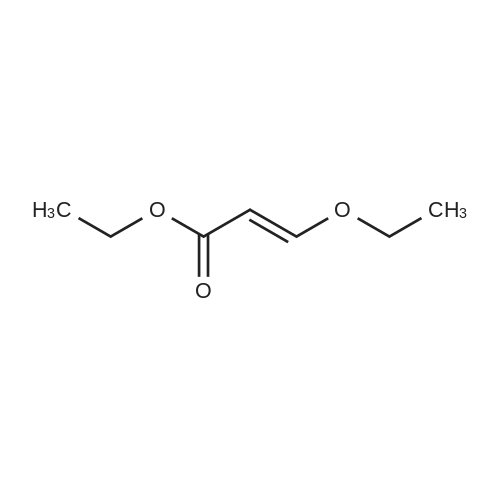
With bis(tri-t-butylphosphine)palladium(0); dicyclohexylmethylamine; lithium chloride; In 1,4-dioxane; at 110℃; for 16h;Inert atmosphere;
General procedure: A flask containing LiCl (980 mg,23.1 mmol), 2-bromo-4-fluoroanisole (1.0 mL, 7.7 mmol), DCMA (1.8 mL,8.4 mmol) and <strong>[1001-26-9]ethyl 3-ethoxyacrylate</strong> (3.3 mL, 23 mmol) in 1,4-dioxane(20 mL) was degassed by passing a stream of nitrogen through the mixturefor 10 min. Bis(tri-t-butylphosphine) palladium(0) (166 mg, 0.32 mmol) wasadded, and reaction mixture was heated at reflux under nitrogen for 16 h. Thebrown mixture was then cooled and partitioned between ethyl acetate andwater. The layers were separated, and the organic layer was washedsequentially with aqueous NH4Cl and brine, followed by drying over Na2SO4.The mixture was filtered, and the filtrate was concentrated under reducedpressure to give an oil which was purified by flash chromatography on silica(40 g, 10?50percent ethyl acetate in heptane) to afford 22c as an orange oil (1.93 g,92percent) as an inseparable mixture of E- and Z-isomers: 1H NMR (CDCl3, 500 MHz)alkene protons d: 5.21, 5.33 ppm (1:1 ratio); LCMS (ES): 269.2 (MH). HRMSCalcd for C14H17FO: 269.1184. Found: 269.1196.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping