| 81% |
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 5h; |
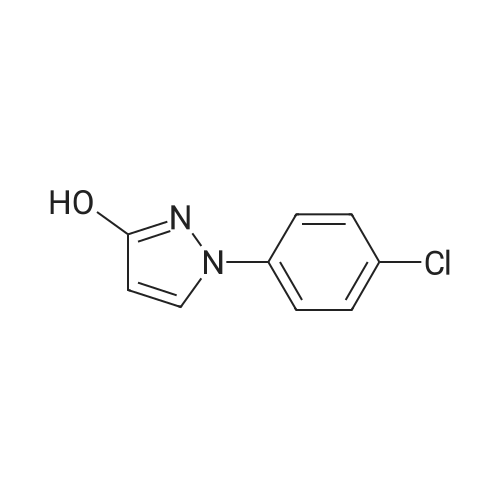
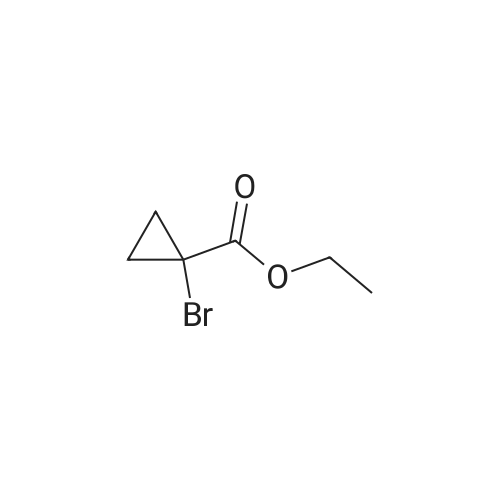
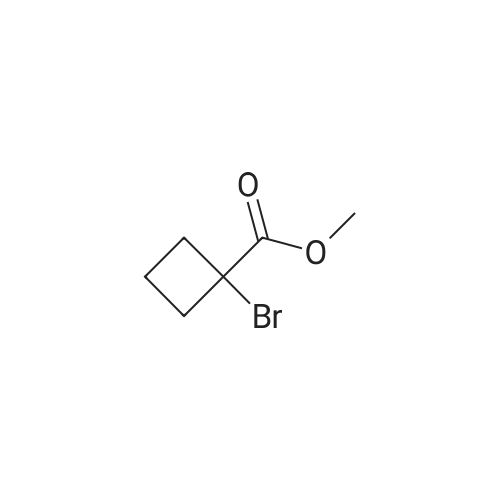
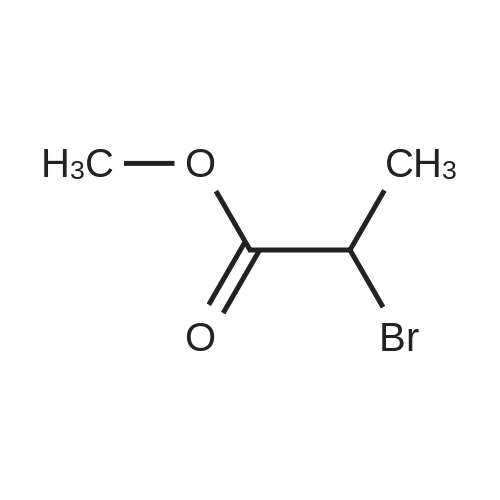
General procedure: The mixture of ethyl 2-bromopropanoate (11.00, 60.0 mmol), B/B? (9.93 g, 50.0 mmol) and potassium carbonate (5.60 g, 40.0 mmol) in N,N-dimethylformamide (DMF; 100 mL) was stirred at 100 C for about 5 h. After completion of the reaction (TLC), the reaction mixture was filtered. The filtrate was evaporated in vacuum to remove DMF and the residue was dissolved in ethyl acetate (50 mL). The organic phase was washed with water (20 mL), evaporated, and the crude product was further purified by recrystallization from ethanol to obtain D1: 12.06 g, yield 81.0%. White solid, m.p. 77-79 C. 1H NMR (300 MHz, CDCl3, 25 C, TMS): delta (ppm) 7.69 (d, J=3.0 Hz, 1H, pyrazol-5-H), 7.48 (d, J=8.7 Hz, 2H, 4-Cl-Ph-2,6-2H), 7.33 (d, J=8.7 Hz, 2H, 4-Cl-Ph-3,5-2H), 5.88 (d, J=2.7 Hz, 1H, pyrazol-4-H), 5.14 (q, J=6.9 Hz, 1H, CH), 4.23 (q, J=7.2 Hz, 2H, CH2), 1.63 (d, J=6.9 Hz, 3H, CH3), 1.26 (t, J=7.2 Hz, 3H, CH3); 13C NMR (75 MHz, CDCl3, 25 C, TMS): delta (ppm) 172.1, 163.0, 138.4, 130.4, 129.2, 127.6, 118.5, 94.5, 72.9, 61.0, 17.9, 14.1; MS (ESI+) Calcd for C14H15ClN2O3 (M+H)+, m/z 295.08, found, 295.1; Anal. Calcd for C14H15ClN2O3: C, 57.05, H, 5.13, N, 9.50, found, C, 57.00, H, 5.12, N, 9.52%. |
| 81% |
With potassium carbonate; In N,N-dimethyl-formamide; at 90 - 100℃; for 5h; |
1-(4-chlorophenyl)-3-pyrazol9.9g (60mmol),Ethyl 2-bromopropionate11.0 g (60 mmol),Potassium carbonate 5.6g (40mmol)And 100 ml of N,N-dimethylformamide was put into a 250 mL three-neck bottle.The temperature was raised to 90 to 100 C and maintained for 5 hours.Thin layer chromatography (TLC) followed the progress of the reaction (n-hexane / ethyl acetate = 4:1). After the conversion of 1-(4-chlorophenyl)-3-pyrazol was completed, the material was filtered to remove the salt, and the filter cake was washed with 20 ml of fresh N,N-dimethylformamide. The filtrate was combined, and the solvent was removed by evaporation under reduced pressure. The residue was washed with 50 g of ethyl acetate and 20 g of water, and the oil phase was distilled under reduced pressure to remove solvent. The residue was recrystallized from ethanol,A solid of 12.0 g was obtained in a yield of 81.0% (based on 1-(4-chlorophenyl)-3-pyrazol). |
| 81% |
With potassium carbonate; In N,N-dimethyl-formamide; at 90 - 100℃; for 5h; |
9.93 g (50 mmol) of <strong>[76205-19-1]1-(4-chlorophenyl)-3-pyrazolyl-ol</strong>, 11.00 g (60 mmol) of ethyl 2-bromopropionate, 5.60 g (40 mol) of potassium carbonate, and100 ml of N,N-dimethylformamide was placed in a 250 mL three-necked flask, and the temperature was raised to 90 to 100 C for 5 hours. Thin layer chromatography (TLC) followed the progress of the reaction (n-hexane / ethyl acetate = 4:1). After the conversion of <strong>[76205-19-1]1-(4-chlorophenyl)-3-pyrazolyl-ol</strong> was completed, the material was filtered to remove the salt and the filter cake was washed with 20 ml of fresh N,N-dimethylformamide. The filtrate was combined, and the solvent was removed by evaporation under reduced pressure. The residue was dissolved in 50 mL of ethyl acetate, washed with water, an oil phase was distilled under reduced pressure. The residue was recrystallized from ethanol to give a white solid 12.06 g. Yield 81.0% (based on <strong>[76205-19-1]1-(4-chlorophenyl)-3-pyrazolyl-ol</strong>). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +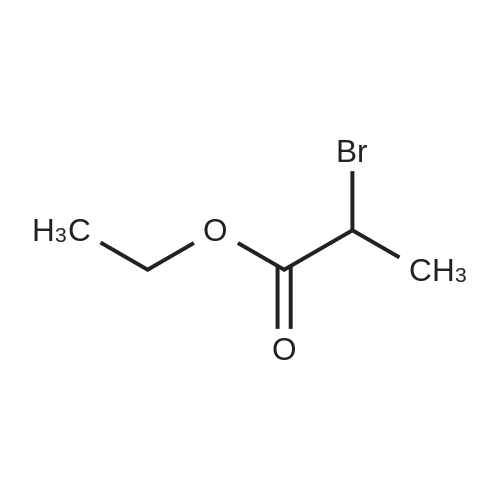

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping