* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With potassium carbonate In acetone at 20℃; for 18 h; Reflux
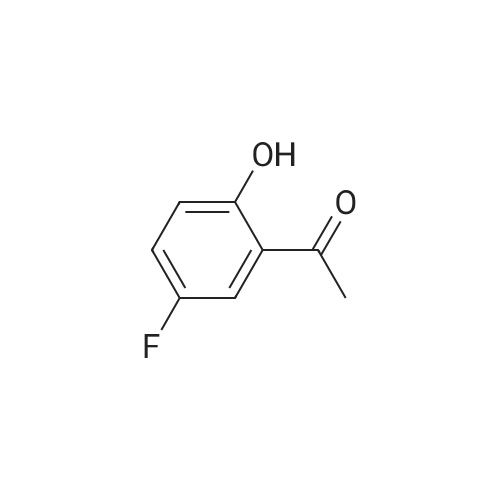
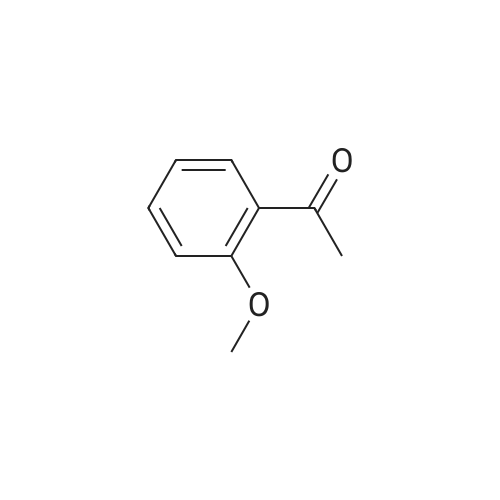
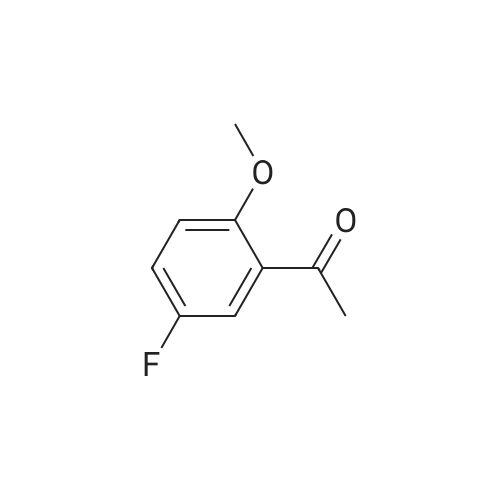
[00217] Step 2. The 4.1 : 1 mixture of compound 10 (10.4 g, 61.8 mmol) and compound 11 (2.54 g, 16.5 mmol) was dissolved in acetone (50 mL) and potassium carbonate (2.50 g, 18.1 mmol) and dimethyl sulfate (0.25 mL, 2.6 mmol) were added. The reaction was refluxed for 18 hours, cooled to room temperature, and water (20 mL) added. This mixture was stirred at room temperature for 3 hours and partitioned between dichloromethane and brine (50 mL each). The layers were separated and the aqueous layer was extracted with dichloromethane (3 x 50 mL). The organics were combined, dried with anhydrous sodium sulfate, and concentrated. This provided 13.0 g (98percent yield, 97 areapercent) of compound 10 as a yellow oil. NMR (400 MHz, CDCh) δ ppm 2.64 (s, 3 H) 3.92 (s, 3 H) 6.94 (dd, J=9.09, 4.04 Hz, 1H) 7.18 (ddd, J=9.09, 7.33, 3.28 Hz, 1H) 7.48 (dd, J=8.97, 3.16 Hz, 1H); HPLC Retention Time: 3.39 min; MS (ESI+) for C9H9FO2 m/z 169.1 (M+H)+.
Reference:
[1] Patent: WO2018/161008, 2018, A1, . Location in patent: Paragraph 00217
[2] Journal of Organic Chemistry, 1960, vol. 25, p. 1016 - 1020
[3] Patent: US4251546, 1981, A,
2
[ 394-32-1 ]
[ 74-88-4 ]
[ 445-82-9 ]
Yield
Reaction Conditions
Operation in experiment
93%
With potassium carbonate In N,N-dimethyl-formamide at 20℃;
General procedure: A mixture of 5-Bromo-2-hydroxyacetophenone 2a (10g, 46.5mmol, 1 eq), potassium carbonate (9.62g, 69.75mmol, 1.5 eq) and iodomethane (5.8mL, 93mmol, 2 eq) in DMF (120mL) were stirred overnight in a sealed round bottom flask at room temperature. DMF was removed under reduced pression and the residue was partitioned between water and EtOAc. The combined EtOAc extracts were washed with brine, 0.5M NaOH and then three times with water. The organic layer was dried over MgSO4 and the solvent evaporated. The product was obtained as an off-white solid (9.5g, 89percent).
Reference:
[1] European Journal of Medicinal Chemistry, 2018, vol. 144, p. 774 - 796
[2] Journal of the American Chemical Society, 2004, vol. 126, # 11, p. 3488 - 3495
3
[ 459-60-9 ]
[ 75-36-5 ]
[ 445-82-9 ]
Yield
Reaction Conditions
Operation in experiment
89%
With aluminum (III) chloride In dichloromethane at 1 - 5℃; for 19 h; Inert atmosphere
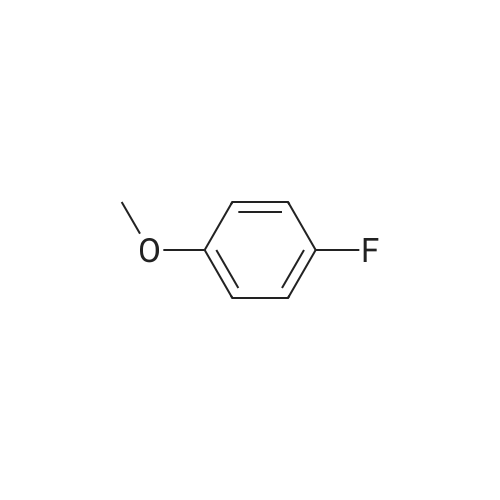
[00206] Example 3.9: Two-Step Preparation of 2-chloro-l-(5-fluoro-2- methoxyphenyl)ethanone (compound 1) [00207] Compound 1 in Scheme 4 was generally prepared in a two-step procedure according to Scheme 5 below.Scheme 5: Synthetic scheme for the preparation of Compound 1 of Scheme 4 [00208] Step 1. A 125-mL three-necked jacketed reaction flask fitted with a temperature probe and nitrogen balloon was charged with aluminum chloride (12.7 g, 95.1 mmol) and dichloromethane (50 mL). This mixture was cooled to 1-2 °C and 4-fluoroanisole (compound 9, 8.98 mL, 79.3 mmol) was added slowly over a period of 30 minutes to maintain the temperature below 5 °C. After the mixture had re-cooled to 1-2 °C, neat acetyl chloride (7.89 mL, 1 1 1 mmol) was added dropwise over a period of 30 minutes, maintaining the temperature below 5 °C. The reaction was then allowed to stir at 1-2 °C for 18 hours. [00209] A 1-L three-necked round bottom flask fitted with a temperature probe and mechanical stirrer was charged with sodium hydroxide (20.2 g, 504 mmol) and water (200 mL) followed by the slow addition of acetic acid (28.7 mL, 504 mmol) while cooling in an ice bath. The homogeneous Friedel-Crafts mixture was diluted with dichloromethane (25 mL) and slowly added dropwise via cannula to this cold (0-5 °C) solution of sodium acetate at a rate that maintained the temperature below 8 °C. Dichloromethane (100 mL) was added after the addition was complete and the mixture allowed to warm to room temperature and stirred for 60 minutes. This solution was transferred to an addition funnel, the layers were separated and the aqueous layer was extracted with dichloromethane (2 x 100 mL). The organics were combined, washed with IN NaOH (3 x 75 mL), dried with anhydrous sodium sulfate and concentrated. This provided 11.9g (89percent yield, 96 areapercent) of compound 10 as an oil. NMR (400 MHz, CDCh) δ ppm 2.64 (s, 3 H) 3.92 (s, 3 H) 6.94 (dd, J=9.09, 4.04 Hz, 1H) 7.18 (ddd, J=9.09, 7.33, 3.28 Hz, 1H) 7.48 (dd, J=8.97, 3.16 Hz, 1H); HPLC Retention Time: 3.39 min; MS (ESI+) for C9H9FO2 m/z 169.1 (M+H)+.
Reference:
[1] Journal of Organic Chemistry, 1954, vol. 19, p. 1617,1621[2] Bulletin de la Societe Chimique de France, 1956, p. 629,632
[3] Patent: WO2018/161008, 2018, A1, . Location in patent: Paragraph 00211-00216
5
[ 579-74-8 ]
[ 445-82-9 ]
Reference:
[1] Chemical Communications, 2000, # 14, p. 1323 - 1324
6
[ 75-16-1 ]
[ 189628-38-4 ]
[ 445-82-9 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 1, p. 619 - 622






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping