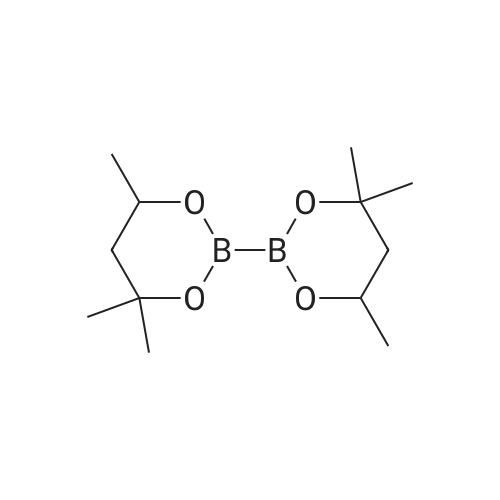
Abstract: In this study, we developed a simple transition-metal-free borylation reaction of aryl bromides. Bis-boronic acid (BBA), was used, and the borylation reaction was performed using a simple procedure at a mild temperature. Under mild conditions, aryl bromides were converted to arylboronic acids directly without any deprotection steps and purified by conversion to trifluoroborate salts. The functional group tolerance was considerably high. The mechanism study suggested that this borylation reaction proceeds via a radical pathway.
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
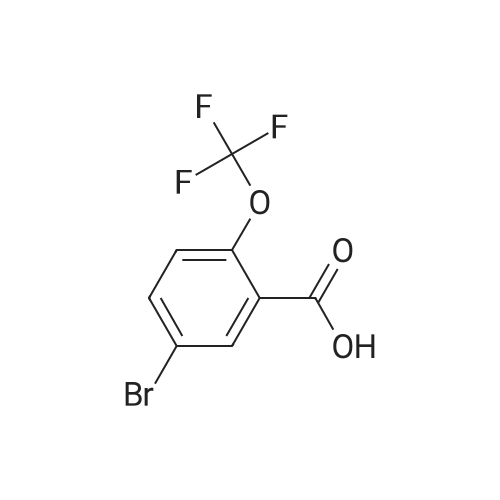
With lithium diisopropyl amide; In tetrahydrofuran; at -78℃; for 2h;
Under argon a solution of LDA (12.45 ml, 2M in THF) was cooled to -78C and a solution of 1-Bromo-4-trifluoromethoxy-benzene (3.7 ml) in THF (50 ml) was added slowly. The reaction mixture was stirred at -78C for two hours and dry ice was added in excess. The solvent was distilled off and the crude product was dissolved in aqueous NaOH (1N), extracted with dichloromethane, then acidified with HCl and again extracted with dichloromethane. The organic layers were dried over sodium sulphate and the solvent was removed in vacuum. The title compound was obtained in 49% yield. 1H-NMR (DMSO-d6): 13.78 s (1H); 8.05 d (J = 2.6 Hz, 1H); 7.92 dd (J = 2.6 Hz / 8.9 Hz, 1H); 7.47 dd (J = 1.3 Hz / 8.9, 1H).
49%
2b) 5-Bromo-2-trifluoromethoxy-benzoic acid Under argon a solution of LDA (12.45 ml, 2M in THF) was cooled to -78C and a solution of 1-Bromo-4-trifluoromethoxy-benzene (3.7 ml) in THF (50 ml) was added slowly. The reaction mixture was stirred at -78C for two hours and dry ice was added in excess. The solvent was distilled off and the crude product was dissolved in aqueous NaOH (1 N), extracted with dichloromethane, then acidified with HCl and again extracted with dichloromethane. The organic layers were dried over sodium sulphate and the solvent was removed in vacuum. The title compound was obtained in 49% yield. 1H-NMR (DMSO-d6): 13.78 s (1 H); 8.05 d (J = 2.6 Hz, 1 H); 7.92 dd (J = 2.6 Hz / 8.9 Hz, 1 H); 7.47 dd (J = 1.3 Hz / 8.9, 1 H).
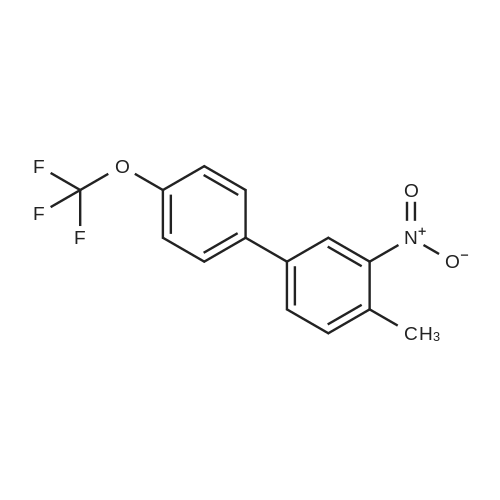
With tetrabutylammomium bromide; palladium diacetate; sodium carbonate; In water; at 150℃;Microwave irradiation; Sealed vessel;
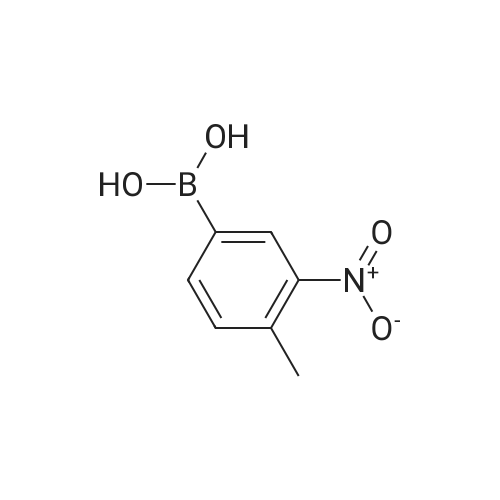
General procedure: Method A. A solution of Pd(OAc)2 (25.2 mg, 0.112 mmol) and triphenylphosphine (147 mg, 0.560 mmol) in absolute ethanol (4 mL) and anhydrous toluene (4 mL) was stirred at RT under nitrogen for 10 min. After that period, commercially available 5-chloro-2-nitrotoluene 4 (646 mg, 3.76 mmol), 4 mL of 2M aqueous Na2CO3, and the appropriate boronic acid R1B(OH)2 (6.03 mmol) were sequentially added. The resulting mixture was heated at 100 C in a sealed vial under nitrogen overnight. After being cooled to RT, the mixture was diluted with water and extracted with EtOAc. The combined organic phase were dried and concentrated. The crude product was purified by flash chromatography over silica gel column using n-Hex/EtOAc or CHCl3/MeOH mixtures as the eluent.
tetrahydro-2H-pyran-4-yl(4-(trifluoromethoxy)phenyl)methanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
812 mg
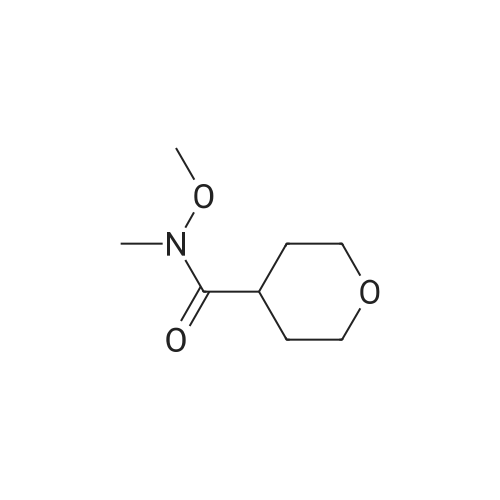
B) tetrahydro-2H-pyran-4-yl(4-(trifluoromethoxy)phenyl)methanone To a solution of 1-bromo-4-(trifluoromethoxy)benzene (2.78 g) in tetrahydrofuran (76.8 mL) was slowly added 1.6 M n-butyllithium/hexane solution (7.20 mL) at -78C. The reaction mixture was stirred at -78C for 30 min under nitrogen atmosphere, a solution of <strong>[156353-01-4]N-methoxy-N-methyltetrahydro-2H-pyran-4-carboxamide</strong> (665 mg) in tetrahydrofuran (1.00 mL) was added thereto at -78C. The reaction mixture was stirred at room temperature for 2 hr under nitrogen atmosphere, saturated aqueous ammonium chloride solution was added thereto, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/ethyl acetate) to give the title compound (812 mg). MS (API+): [M+H]+275.1.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping