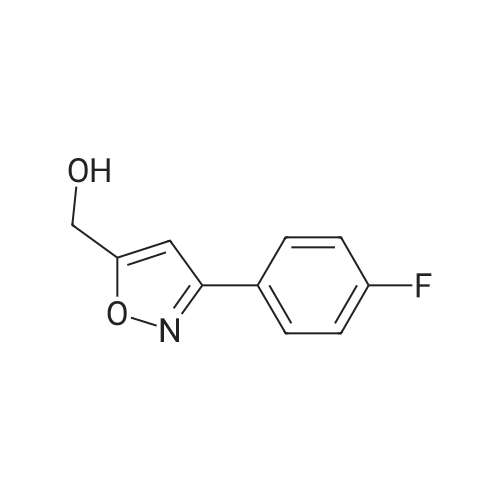
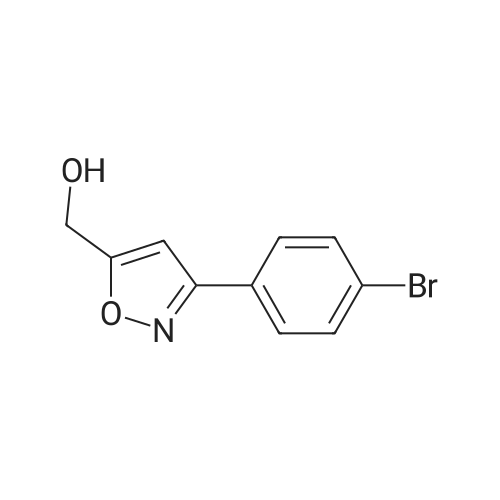
|
With sulfonic chloride acid; In dichloromethane; at -5 - 35℃; for 10h; |
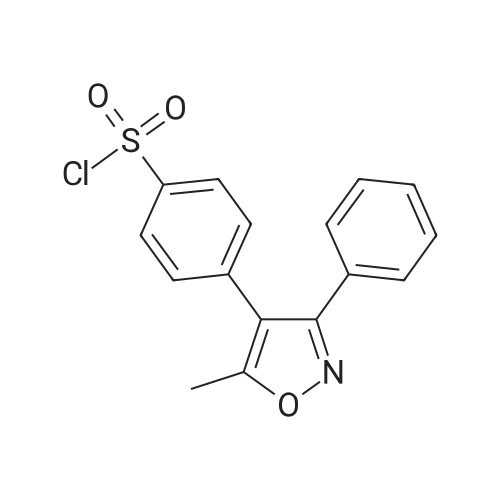
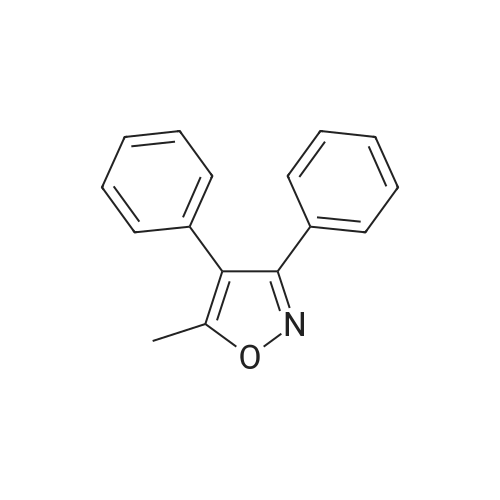
To the reaction flask, 8.0 g of 5-methyl-3,4-diphenylisoxazole and 40 g of methylene chloride were added and the temperature was lowered to -5 C in an ice bath. 32 g of chlorosulfonic acid was added dropwise, and the temperature of the reaction solution was controlled to <5 Lt; 0 & gt; C. After the dropwise addition of chlorosulfonic acid, the temperature was slowly raised to 35 C and the reaction was allowed to proceed for 10 h. The reaction mixture was stopped by monitoring the reaction mixture to 5-methyl-3,4-diphenylisoxazole (the developing solvent was ethyl acetate: petroleum ether = 1: 6, v: v). 100g of crushed ice was added to the reaction flask, stirred for 1h, poured into a separatory funnel and allowed to stand for separation, and the lower aqueous phase was separated. The upper organic phase by adding 5g anhydrous sodium sulfate drying 2h, filtered to get a clear liquid. The organic phase was concentrated in vacuo. The concentration was 45 C and the vacuum was -0.08 MPa. And concentrated to dryness to give a pale yellow solid as the main component,The reaction mixture was heated to 75 C for 6 hours, and the reaction was monitored by TLC until the spots disappeared in the intermediate I (developing solvent: ethyl acetate: petroleum ether = 1: 1) and the mixture was stirred for 1 hour. 1: 8, v: v) to stop the reaction. Hot vacuum concentrated ethanol about 25ml, cooling, stirring crystallization at -5 above 6h. Filtration, crude products were 13.5g tide.13.5 g of the above crude product was added to 100 ml of water, stirred at a temperature of 25 C for 2 hours, and then filtered. Filtration products directly after the addition of ethanol 30ml, heated to 75 reflux to the total solution, the cooling, stirring crystallization at -5 above 6h. Filtration, in refined products tide goods. Dried at 45 C for 8 h. The final product, ethyl 4- (5-methyl-3-phenyl-isoxazolyl) benzenesulfonate, was obtained in a yield of 61.7% |
|
With sulfonic chloride acid; In dichloromethane; at -5 - 35℃;Large scale; |
The 3.0 kg 5 - methyl - 3, 4 - diphenyl isoxazole added to the 30 L glass reaction flask, then adding 9.9 kg methylene chloride, stirring to dissolve, cooling the solution to - 5 - 0 C, to the reaction solution slowly dropping 15.0 kg chloro sulfonic acid and temperature control ≤ 20 C, after dropping, water bath heating to 35 C insulation reaction 4 - 5 of H, then the reaction liquid cooling to 0 - 5 C.The 15.9 kg methylene chloride and 12.0 kg purified water by adding another 50 L in the reaction bottle, cooling to 0 - 5 C, slowly dropping has cooling of the reaction fluid and temperature control ≤ 20 C, after dropping stirring 0.5 h; layered, for methylene chloride level (the upper layer) 12.0 kg (25% w/w) saturated sodium chloride solution agitation washing, the delaminated, taking methylene chloride level (lower) spare; to the 50 L reaction flask add 5.4 kg concentrated ammonia water, then slowly adding the dichloromethane layer solution, at room temperature (20 - 30 C) stirring the reaction 2 - 3 H of the; added to the reaction solution after 11.7 kg isopropanol, 35 C thermal insulation to continue stirring 1.5 h, then cooled to 15 - 20 C, stirring crystallization 6 h, then adding, for material 2.4 kg of cold isopropanol washing, 60 C drying under atmospheric pressure 5 - 10 the H, get-cutting to past cloth thick 3.1 kg; will-cutting to past cloth thick 3.1 kg and 7.36 kg methanol by adding 30 L glass reaction flask, stirring under heating to 60 - 70 C reflux, filtered to remove insoluble matter, then adding 3.1 kg purified water, the temperature slowly drops to 15 - 20 C, stirring crystallization 6 h, filtered, in order to 3.1 kg cold methanol/water (V/V=3:1, W/W=2.37: 1) in the solution, 60 C drying under atmospheric pressure 5 - 10 the H, get-cutting to [...] 2.2 kg. |
| 30 g |
With sulfonic chloride acid; In dichloromethane; at 40℃; |
1) Chlorosulfonic acid (100g),The compound of formula 1-1 (50 g) was added sequentially to 50 mL of dichloromethane.The reaction was carried out at 40 C overnight.2) The reaction solution was monitored by TLC (thin layer chromatography), and a trace amount of raw material remained.3) After cooling to room temperature, the reaction was dropped into ice (about 500 g) and extracted with ethyl acetate (500 mL) to obtain organicphase. The organic phase was concentrated to dryness to give an oil. Add 100 mL of ethyl acetate to the oil, add 600 mL of petroleum ether, and stir.2h, a solid precipitated. The filter cake was washed by filtration with a mixed solvent of petroleum ether / ethyl acetate (6:1). Vacuum drying the filter cakeThe compound of formula 1-3 (30 g) was obtained. |
|
With sulfonic chloride acid; In dichloromethane; at 0 - 60℃; for 5h; |
SM (300 g, 1275 mmol) was added to a 5 L three-necked flask.Mix well in dichloromethane (300 ml ice bath).Temperature control is around 0-5 degrees,Dichloromethanesulfonic acid in dichloromethane (1486 g, 12750 mmol,Methylene chloride (700ml),After 2 hours, the addition is completed.Warm up to 60 degrees and stir for 3 hours.The TLC dot plate showed complete reaction (PE: EA = 4:1).The system was added dropwise to 1200 ml of ice water.Temperature control is about 10 degrees,Liquid separation extraction,The organic phase is washed twice with water.The organic phase is stirred with ammonia (700 ml) at room temperature.The dot plate shows that the reaction is complete,(PE: EA=4:1),The organic phase is removed by a 50 C rotary steaming system.The remaining system was added with 2 L of ethyl acetate.1L of water is stirred until it is dissolved.Liquid separation extraction,The aqueous phase was back extracted with ethyl acetate.Combine the organic phase,Wash with purified water,The organic phase was removed by rotary evaporation to give a crude material.Add 1L of ethanol,Heat to dissolve,Recrystallization,Filtered to give a crude product (350g).The final product 310 g (986.13 mmol) was obtained (HPLC > 99.5%).White solid, yield 77%. |
|
With sulfonic chloride acid; In dichloromethane; at -2 - 35℃; for 0.5h; |
Add 50g of dichloromethane and 5g of starting material 5-methyl-3,4-diphenylisoxazole to a 100mL three-neck reaction flask, stir and cool to -2 C, connect the tail gas adsorption device at a rate of 2mL / min , Slowly add 16.5g of chlorosulfonic acid to control the reaction temperature up to 25 .After the dropwise addition, it was stirred for 0.5 hours, and then heated to 35 C.The reaction progress was monitored by HPLC, calculated by the area normalization method, until the starting material 5-methyl-3,4-diphenylisoxazole≤1.0%, meta-chlorosulfonate isomer≤1.0%, the reaction was stopped , Get the reaction solution.Slowly drop the reaction solution into ice water, cool it, control the maximum temperature not to exceed 15 C, and stir for 0.5 hours after the dropwise addition.The solution was separated by standing, extracted with fresh dichloromethane, and then the organic phases were combined, washed with aqueous sodium chloride solution and separated, and dried over anhydrous sodium sulfate.Suction filtration to obtain a solution of para-chlorosulfonate intermediate in dichloromethane for use. |
|
|
Add 80mL of dichloromethane to the reaction bottle, add compound I11.76g, add formic acid 4.60g, add chlorosulfonic acid 58.26g dropwise at a temperature control of 10-20 C, dropwise finish after 15min, then stir for 15min, add potassium carbonate 27.64g, add Sodium iodide 1.18g, heated to 35-45 C, and the reaction was completed after 1h by TLC (developing agent n-heptane: ethyl acetate = 4: 1, compound I Rf = 0.9, compound II Rf = 0.7). Add the reaction dropwise to the purified water at 0 5 , and control the temperature to be below 20 during the dropwise addition. After the dropwise addition, add 60mL of ethyl acetate and stir for 10min, let stand for 30min, separate the liquid, and wash the organic phase once with purified water. A solution of compound II in ethyl acetate was obtained, and 32.93 g of 25% -28% concentrated ammonia solution was added. The temperature was controlled at 20-25 C and stirred for 2 h. The reaction was monitored by TLC (developing agent n-heptane: ethyl acetate = 2: 1, (Rf of compound II = 0.9, Rf of compound III = 0.4), stand for 30 minutes, separate the layers, wash the organic phase twice with purified water, evaporate the solvent under reduced pressure at 50 C, and add 80 mL of absolute ethanol-methanol (10 : 1), heated to 60 , stirred until dissolved, slowly cooled to 0 ~ 5 , stirred and crystallized for 1h, suction filtered, vacuum dried at 55-60 for 8h, to obtain 14.59g compound III, yield 92.8% , HPLC detection purity 99.8%, the largest single impurity 0.08%. |
|
With sulfonic chloride acid; In dichloromethane; at 40℃; for 5h; |
Add 50g of 5-methyl-3,4-diphenylisoxazole, 250ml of dichloromethane and 198g of chlorosulfonic acid into a reaction flask, and react under reflux at 40C for 5 hours.After the reaction is completed, the temperature is lowered to 05C, and it is added dropwise to 800g ice water and stirred for 30 minutes.Static stratification, liquid separation, 400ml dichloride phase washed once with water, directly cast to the next step. |
| 7.12 kg |
With sulfonic chloride acid; In dichloromethane; at 0℃; for 4h;Reflux; Large scale; |
15.6kg of dichloromethane and 6.0kg of SM1 were added to the reaction kettle, and the temperature was lowered to 0-15C with stirring.Slowly add 26.64kg of chlorosulfonic acid dropwise, and control the internal temperature to 0-15C. dripping,The reaction was heated to 40-45C and refluxed for 4 hours. Cool down to 0-15C,The reaction solution was slowly added dropwise to 72 kg of purified water, and the internal temperature was controlled to 0-30C. static layering,The organic phase was separated, and the aqueous phase was extracted with 18 kg of ethyl acetate (no emulsification, no need to stand),The organic phase was separated and washed with 5.0 kg of purified water. After concentrating the organic phase of dichloromethane, add the organic phase of ethyl acetate and concentrate again until the remaining mass of the system is 15kg±1kg.The temperature was controlled at 30-40C, and 33kg of n-hexane was added dropwise, and the temperature was slowly lowered to 10-20C, stirred and crystallized for 1 hour, centrifuged, rinsed with 6kg of n-hexane, and dried under vacuum at 60-65C to obtain 7.12kg of white solid.The purity is 99.36% (see Figure 1 for details). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping