* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
N-{3-[(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-amino]-4-methoxy-phenyl}-benzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Example 271 N-{3-[(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-amino]-4-methoxy-phenyl}-benzamide The title compound has been made using the procedure of Example 50, but using 3-amino-4-methoxy-N-phenyl benzamide and 3,4-ethylenedioxybenzaldehyde as starting materials, which can be purchased from Aldrich; m.p. 174-175 C.
With ammonia; hydrogen;Raney Nickel; In methanol; at 20 - 50℃; under 2585.81 - 2844.39 Torr;Autoclave;
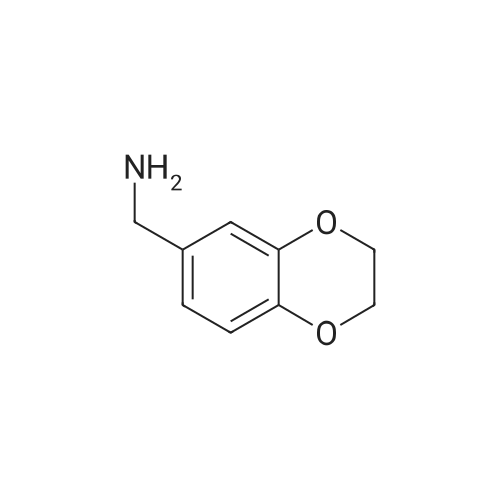
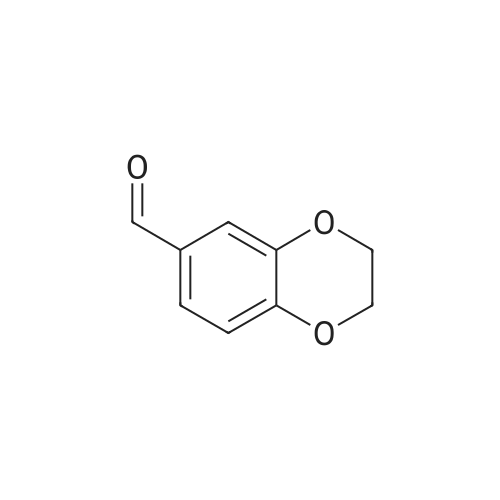
Example-8Preparation of 2,3-dihydro-benzo[1,4]dioxin-6-yl-methylamine hydrochloride 242.0 g of Methanolic ammonia [as 100% w/w by chemical assay] [Note: as chemical assay: 23.0% w/w, volume 1350.0 ml] and 30.0 g (0.183 mol) of 1,4-benzodioxan-6-carboxaldehyde were charged into a 2.0 L 4 necked round bottom flask, connect to a mechanical stirrer, thermo meter socket and condenser at 20-30 C. Stirred the mass for 20-30 min at 20-30 C. After dissolution is clear. Reaction mass was charged into a 2.0 L hydrogenator kettle at 20-30 C. 30.0 g of Raney Nickel (with Methanol dried) was charged under nitrogen atmosphere. Kettle was fitted to the hydrogenator. Nitrogen atmosphere was removed in Hydrogenator kettle with hydrogen gas by slowly flushing. Hydrogen gas was feeded upto 50-55 psi in hydrogenation kettle under oscillation. Maintained the hydrogen gas pressure (50-55 psi) till the hydrogen gas consumption is stopped. Reaction mass temperature was raised to 40-45 C. After hydrogen gas consumption is stooped at 45-50 C. Reaction mass temperature was cooled to 25-30 C. Maintained the hydrogen gas pressure at 50-55 psi for till the hydrogen gas consumption is stopped (about 90-120 min) Raney nickel was filtered through hyflow bed under nitrogen atmosphere. Raney Nickel was washed with 300.0 ml of methanol under nitrogen atmosphere. Filterate was collected into a flask. Methanol was distilled completely under vacuum at mass temperature not crossing 55 C. Mass temperature was cooled to 40-45 C. and release the vacuum. 50.0 ml of isopropyl alcohol was added. Reaction mass pH was adjusted to 0.5+/-0.25 with IPA HCl. Maintained the mass temperature at 25-30 C. for 60-90 min under stirring. Solid was filtered and solid was washed with 20.0 ml of isopropyl alcohol. Compound was dried under vacuum at 40+/-5 C. Dry compound weight: 31.0 g (yield: 84.1%).Spectral data:FT-IR (K Br) (cm-1): 3447.6, 2977.6, 2870.0, 1594.5, 1506.6, 1474.0, 1285.8, 1077.7, 1051. 735.2, 617.9, 472.0MS: 202.6 [M+1]
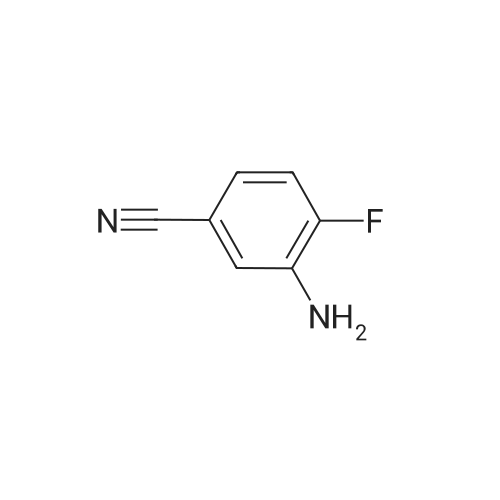
Intermediate 8: 3-[(2,3-dihydro-l,4-benzodioxin-6-ylmethyl)amino]-4-fluorobenzonitrile; A suspension of 1 ,4-benzodioxan-6-carboxaldehyde (ABCR; 1.03 g; 6.27 mmol) and 3-amino-4- fluorobenzonitrile (Intermediate 1; 997 mg; 7.32 mmol) in Toluene (100 ml) was treated with p- toluenesulphonic acid monohydrate (10 mg; 0.06 mmol). A Dean-Stark trap was added and the suspension heated at reflux during 20 h, after which the reaction solution was cooled and concentrated to give a yellow solid. This was dissolved in MeOH (100 ml) and DCM (300 ml). The solution was cooled to -10 0C then treated with three portions OfNaBH4 (441 mg; 11.7 mmol each, 20 minutes apart). After stirring at room temperature for 3 h, the solution was concentrated then dissolved in DCM and washed with aqueous HCl (1 N). The layers were separated and the organic layer dried on MgSO4 and concentrated to give the Title compound (1.88 g, 90%) as an orange oil, which was used without further purification.1H NMR (300MHz, DMSOd6) delta [ppm] 7.27-7.16 (m, IH), 7.02-6.93 (m, 2H), 6.90-6.68 (m, 4H), 4.25 (d, J= 6.3 Hz, 2H), 4.20 (s, 4H). HPFC (Condition A): Rt 4.04 min (HPFC purity 89.2%).
General procedure: In the first synthetic step (step a, Scheme 1), a series of (Z)-substituted diarylacrylonitrile analogues were synthesized by reacting substituted benzyl carbaldehydes with their corresponding substituted phenylacetonitriles in 5% NaOMe in methanol. The reaction mixture was stirred at room temperature for 2-3 h for the reaction to complete and the final product precipitated of the solution. The precipitate was filtered, washed with water and dried to yield the final compound in yields ranging from 70 to 95% (Scheme 1) [16].
With sodium methylate; In methanol;Reflux;
General procedure: the aromatic carbaldehyde (1 mole) and the appropriate substituted phenylacetonitrile (1.1 mol equiv) were added to 5% sodium methoxide in methanol and the mixture was stirred at reflux temperaturefor 2-3 h. The resulting solid was filtered off, washed with water, and finally washed with cold methanol. The obtained crude solid was recrystallized from methanol to afford the desired product as a pure crystalline solid. The characterization data for the active compounds is given in reference section.
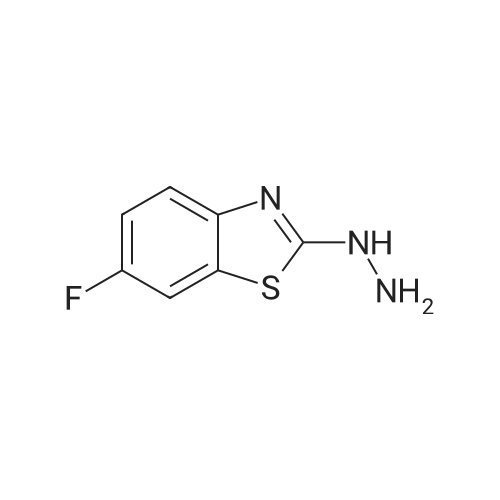
2-[2-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methylene)hydrazino]-6-fluorobenzothiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
81%
With acetic acid; In ethanol; at 80℃; for 0.166667h;Microwave irradiation;
General procedure: 2-(2-Arylidenehydrazino)-6-fluorobenzothiazoles 6a-r. General Procedure D. A mixture of compound 2 (0.0549 g, 0.0003 mol), the appropriate aromatic aldehyde (0.00033 mol) and glacial acetic acid (0.1 mL) in ethanol (5 mL) was heated under microwave (20 W) at 80 °C for 10 min. On cooling, the precipitated solid was collected by filtration, washed with water, dried and crystallized from the appropriate solvent to give the desired compounds 6a-r.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping