| 79% |
With diethylamino-sulfur trifluoride; In dichloromethane; at -78 - 20℃; for 21h; |
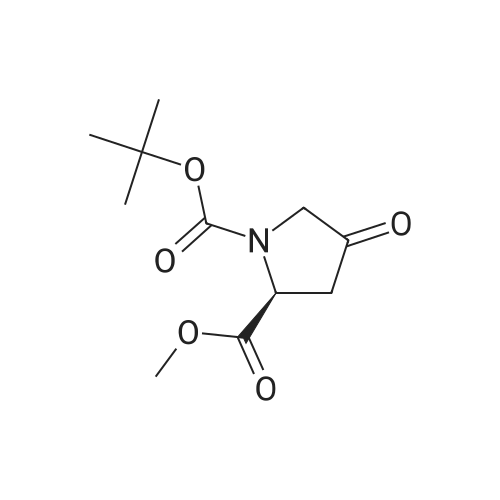
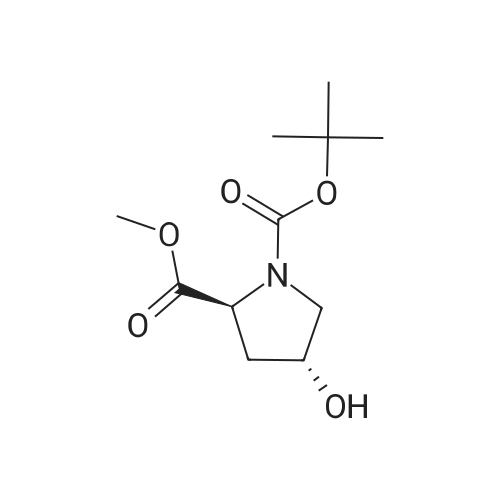
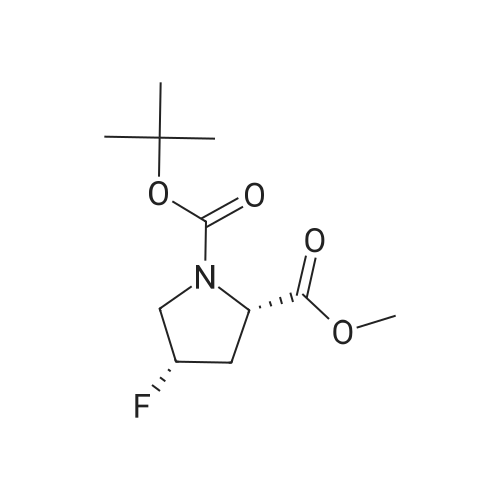
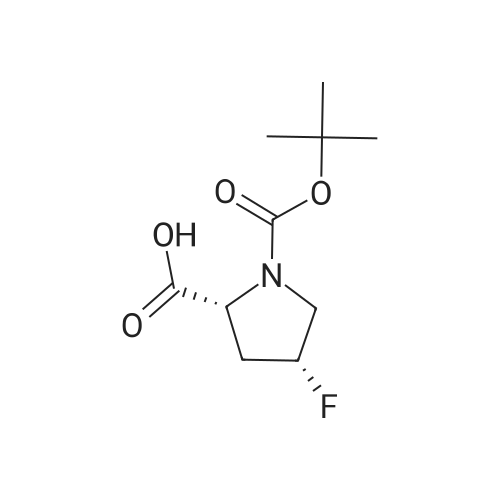
To a solution of compound 14-2 (5.8 g, 23.9 mmol) in DCM (70 mL) at -78 C was added Et2NF3 (4.85 mL, 35.9 mmol) dropwise. At the end of addition, the mixture was stirred at -78 C for 2 hrs and then at rt for another 19 hrs. The reaction was quenched with NH4C1 aqueous solution (50 mL), and the resulting mixture was extracted with DCM (60 mL x 3). The combined organic layers were dried over anhydrous Na2S04 and concentrated in vacuo. The residue was purified by a silica gel column chromatography (PE/EtOAc (v/v) = 20/1 ) to give the title compound as pale yellow liquid (5.0 g, 79%). The compound was characterized by the following spectroscopic data: MS (ESI. pos.ion) ml . 266.25 [M+H] +; NMR (400 MHz, CDC13) delta (ppm): 4.68-4.63 (m, 1 H). 4.01 -3.87 (m. 1 H), 3.78 (s. 3H), 3.75-3.63 (m, 1H). 2.84-2.66 (m. 1 H), 2.51 -2.31 (m. 1H). 1.43 (d. 9H, J =16 Hz). |
| 79% |
With diethylamino-sulfur trifluoride; In dichloromethane; at -78 - 20℃; for 21h;Inert atmosphere; Sealed tube; |
To a solution of compound 12-1 (5.81 g, 23.9 mmol) in DCM (70 mL) was added Et2NSF3 (4.85 mL, 35.9 mmol) dropwise at -78 C. At the end of the addition, the mixture was stirred at -78 C for 2.0 hrs and at rt for another 19 hrs. After the reaction was completed, the mixture was quenched with NH4C1 aqueous solution (50 mL). The aqueous layer was extracted with DCM (100 mL x 3). The combined organic layers were dried over anhydrous Na2S04 and concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EtOAc (v/v) = 20/1) to give the title compound as pale yellow liquid (5.0 g, 79%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) mlz: 266.2 [M+H]+; and NMR (400 MHz, CDC13) delta (ppm): 9.60 (brs, 1H), 4.60-4.57, 4.94-4.72 (m, m, 1H), 3.93-3.84 (m, 2H), 3.77 (s, 3H), 2.78-2.48 (m, 2H), 1.44 (d, 9H, J= 16 Hz). |
| 79% |
With diethylamino-sulfur trifluoride; In dichloromethane; at -78 - 20℃; for 21h; |
To a solution of compound 12-6 (5.81 g, 23.9 mmol) in DCM (70.0 mL) was added Et2NF3(4.85 mL, 35.9 mmol) dropwise at -78 C. At the end of the addition, the mixture was stirred at -78 C for 2.0 hrs and then at rt for another 19 hrs. After the reaction was completed, the mixture was quenched with NH4C1 aqueous solution (50 mL), and the resulting mixture was extracted with DCM (60 mL x 3). The combined organic layers were washed with brine, dried over anhydrous Na2S0 and concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EtOAc (v/v) = 20/1) to give the title compound as pale yellow liquid (5.0 g, 79%). The compound was characterized by the following spectroscopic data:MS (ESI. pos.ion) mlz: 266.25 [M+H]': and NMR (400 MHz, CDCh) 6 (ppm): 4.68-4.63 (m. 1H), 4.01-3.87 (m, 1H).3.78 (s.3H), 3.75-3.63 (in. 1H), 2.84-2.66 (m, 1 H).2.51-2.31 (m.1H).1.43 (d, 9H..7 = 16 Hz). |
| 79% |
With diethylamino-sulfur trifluoride; In dichloromethane; at -78 - 20℃; for 14h; |
To a solution of compound 15-2 (0.5 g, 2 mmol) in DCM (10 mL) at -78 C was added dropwise Et2NSF3 (0.82 mL, 6 mmol). The mixture was stirred at -78 C for 2 hours and at rt for another 12 hours. A small amount of water was added to the mixture, and the resulting mixture was extracted with DCM (30 mL x 3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EtOAc (v/v) = 9/1) to give the title compound 15-3 (0.42 g, 79%). The compound was characterized by the following spectroscopic data: MS-ESI: m/z 266.25 [M+H]+; and1H NMR (400 MHz, CDCl3): delta 4.43-4.47 and 4.55-4.53 (m, 1H), 3.80-3.83 (m, 2H), 3.77 (s, 3H), 2.66-2.74 (m, 1H), 2.40-2.52 (m, 1H), 1.42-1.47 (d, J = 20 Hz, 9H). |
| 79% |
With diethylamino-sulfur trifluoride; In dichloromethane; at -78 - 20℃; for 21h; |
To a solution of compound 5-9 (5.81 g, 23.9 mmol) in DCM (70 mL) at -78 C was added Et2NSF3 (4.85 mL, 35.9 mmol) dropwise slowly. At the end of the addition, the mixture was stirred for 2 hours at -78 C and then stirred at rt for 19 hours. After the reaction was completed, the reaction was quenched with an aqueous solution of NH4CI (50 mL) and the aqueous layer was extracted with DCM (100 mL x 3). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EtOAc (v/v) = 20/1) to give the title compound as pale yellow liquid (5.0 g, 79%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) mlz: 266.3 [M+H]+; and H NMR (400 MHz, CDC13): delta 9.60 (brs, IH), 4.60-4.57, 4.94-4.72 (m, m, IH), 3.93-3.84 (m, 2H), 3.77 (s, 3H), 2.78-2.48 (m, 2H), 1.44 (d, 9H, J= 16 Hz) ppm. |
| 79% |
With diethylamino-sulfur trifluoride; In dichloromethane; at -78 - 20℃; for 21h; |
11121] To a solution of compound 14-2 (5.8 g, 23.9 mmol) in DCM (70 mE) at -78C. was added Et2NF3 (4.85 mE, 35.9 mmol) dropwise. At the end of addition, the mixture was stirred at -78C. for 2 irs and then at it for another 19 hrs. The reaction was quenched with NH4C1 aqueous solution (50 mE), and the resulting mixture was extracted with DCM (60 mEx3). The combined organic layers were dried over anhydrous Na2504 and concentrated in vacuo. The residue was purified by a silica gel column chromatography (PE/EtOAc (v/v)=20/1) to give the title compound as pale yellow liquid (5.0 g, 79%). The compound was characterized by the following spectroscopic data:11122] MS (ESI, pos.ion) mlz: 266.25 [M+H]11123] ?H NMR (400 MHz, CDC13) oe (ppm): 4.68-4.63 (m, 1H), 4.01-3.87 (m, 1H), 3.78 (s, 3H), 3.75-3.63 (m, 1H),2.84-2.66 (m, 1H), 2.51-2.31 (m, 1H), 1.43 (d, 9H, J=16 Hz). |
| 79% |
With diethylamino-sulfur trifluoride; In dichloromethane; at -78 - 20℃; for 14h; |
To a solution of compound 15-2 (0.5 g, 2 mmol) in DCM (10 mL) at -78 C was added dropwise Et2NSF3 (0.82mL, 6 mmol). The mixture was stirred at -78 C for 2 hours and at rt for another 12 hours. A small amount of water wasadded to the mixture, and the resulting mixture was extracted with DCM (30 mL x 3). The combined organic phaseswere dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography(PE/EtOAc (v/v) = 9/1) to give the title compound 15-3 (0.42 g, 79%). The compound was characterized bythe following spectroscopic data:MS-ESI: m/z 266.25 [M+H]+; and1H NMR (400 MHz, CDCl3): delta 4.43-4.47 and 4.55-4.53 (m, 1H), 3.80-3.83 (m, 2H), 3.77 (s, 3H), 2.66-2.74 (m, 1H),2.40-2.52 (m, 1H), 1.42-1.47 (d, J = 20 Hz, 9H). |
| 79% |
With diethylamino-sulfur trifluoride; In dichloromethane; at -78 - 20℃; for 21h; |
Compound 26-3 (5.81 g, 23.9 mmol) was dissolved in DCM (70 mL) and Et 2NSF 3(4.85mL, 35.9 mmol) was slowly added dropwise to the system. After the addition wascompleted, the reaction was carried out at a constant temperature for 2.0 hours and atroom temperature for 19 hours. After the reaction was completed, the reaction wasquenched with aqueous ammonium chloride (50 mL) and the aqueous layer was extracted withDCM (100 mL x 3). The combined organic phases were dried over anhydrous Na 2SO 4Dried andconcentrated. The residue was purified by column chromatography (eluent: PE / EtOAc (v /v) = 20/1) to give 5.0 g of a pale yellow liquid. Yield: 79%. |
| 73% |
With diethylamino-sulfur trifluoride; In dichloromethane; at 0 - 20℃; |
Step 2 Preparation of (S)-1-tert-butyl 2-methyl 4,4-difluoropyrrolidine-1,2-dicarboxylate To a stirred solution of (S)-1-tert-butyl 2-methyl 4-oxopyrrolidine-1,2-dicarboxylate (2.9 g, 11.9 mmol) in DCM (20 mL) was added DAST (5.76 g, 35.8 mmol) dropwisely at 0 C. After being stirred at r.t. overnight, the mixture was quenched with water (10 mL) at 0 C., pH was adjusted to 7?8, extracted with DCM (50 mL*3). The organic layers were washed with brine, dried over Na2SO4, filtered, concentrated in vacuo and the residue was purified by column chromatography on silica gel (petroleum ether:EtOAc=10:1) to afford (S)-1-tert-butyl 2-methyl 4,4-difluoropyrrolidine-1,2-dicarboxylate (2.3 g, 73%) as colorless oil. |
| 61% |
With ethanol; diethylamino-sulfur trifluoride; In dichloromethane; at 20℃; for 18h;Inert atmosphere; |
A solution of the (S)-1-tert-butyl 2-methyl 4-oxopyrrolidine-1,2-dicarboxylate (0.23 g, 0.946 mmol), obtained from step 1, in CH2Cl2 (3.0 mL), in a 25-mL flask equipped with a N2 inlet tube and stirring bar, was treated with a solution of diethylaminosulfur trifluoride (0.197 ml, 1.607 mmol) in CH2Cl2 (2.0 mL) at room temperature. Ethanol (0.011 ml, 0.189 mmol) was added (for in situ generation of catalytic quantities of HF) and the mixture was stirred for 18 h at room temperature. The solution was poured into saturated sodium bicarbonate and after CO2 evolution ceased it was extracted into CH2Cl2 (3*15 mL), dried (Na2SO4), filtered, and evaporated in vacuo. Chromatography on silica gel in DCM afforded a yellowish oil. Yield: 0.150 g, 61%. 1H NMR (400 MHz, CDCl3): delta 1.44 (br s, 9H), 2.45 (dq, J=13.6, 5.2 Hz, 1H), 2.61-2.81 (m, 1H), 3.75 (s, 3H), 3.60-3.90 (m, 2H), 4.45-4.55 (m, 1H). MS (ESI) m/z 266.1 [M+H]+ |
| 61% |
With diethylamino-sulfur trifluoride; In dichloromethane; at 0 - 20℃; |
Example 1 Synthesis of (S)-N-(2-(2-cyano-4,4-difluoropyrrolidin-1-yl)-2-oxoethyl)-1-oxo-1,2-dihydroisoquinoline-4-carboxamide Compound 1a: To a stirred solution of 1-(tert-butyl) 2-methyl (S)-4-oxopyrrolidine-1,2-dicarboxylate (1.9 g, 7.36 mmol, 1.0 equiv.), in DCM (15 mL), was added DAST (2.6 ml, 19.85 mmol, 2.6 equiv) drop wise at 0 C. over a period of 30 min, the reaction mixture was stirred at RT for overnight. Progress of the reaction was monitored by NMR. Water (50 mL) was added to the reaction mixture, stirred for 5 min and extracted with DCM (50 mL*3). Combined organic layer was washed with saturated sodium bicarbonate solution (100 mL), brine (100 mL), dried over anhydrous sodium sulphate and concentrated under reduced pressure to obtain 1-(tert-butyl) 2-methyl (S)-4,4-difluoropyrrolidine-1,2-dicarboxylate (1.25 g, 61% Yield) as a red oil. 1H NMR (400 MHz, DMSO-d6) delta 4.41-4.53 (m, 1H), 3.65-3.84 (m, 5H), 2.82-3.01 (m, 2H), 1.41 (s, 4H), 1.35 (s, 5H). |
|
With (bis-(2-methoxyethyl)amino)sulfur trufluoride; In chloroform; at 0 - 20℃; for 19h; |
36.0 g of [bis(2-methoxyethyl)amino]sulfur trifluoride was added dropwise over a period of 5 minutes under ice cooling to a 150 mL chloroform solution of 18.0 g of the compound obtained in step 3-1, after which the reaction mixture was stirred for 19 hours at room temperature. The reaction solution was added dropwise over a period of 10 minutes under ice cooling to 300 mL of a saturated potassium carbonate aqueous solution. After liquid separation, the aqueous layer was extracted with chloroform (30 mL × 2), and the combined organic layer was washed with 30 mL of saturated brine and dried with magnesium sulfate, and the drying agent was filtered off, after which the solvent was distilled off under reduced pressure. The residue thus obtained was purified by column chromatography (silica gel 60, mobile phase: ethyl acetate/n-hexane = 1/4; v/v) to obtain 15.4 g of the titled compound (yellow oily substance). MS (ESI pos.) m/z : 288([M+Na]+) 1 H-NMR (300 MHz, CDCl3) delta (ppm) ; 1.43 & 1.46 (each-s, 9 H), 2.36 - 2.56 (m, 1 H), 2.58 - 2.82 (m, 1 H), 3.69 - 3.92 (m, 2 H), 3.77 (s, 3 H) 4.40 - 4.61 (m, 1 H) |
|
With diethylamino-sulfur trifluoride; In dichloromethane; at -78℃; |
Step B: Diethylaminosulfur trifluoride (1.88 mL, 14.2 mmol) was added to a -780C solution of l-tert-buty 2-methyl (25)-4-oxopyrrolidine-l,2-dicarboxylate (1.73 g, 7.1 mmol) in dichloromethane (20 mL). After stirring at ambient temperature overnight, the reaction mixture was concentrated in vacuo. Chromatography over silica eluting with 0-50% ethyl acetate/hexane afforded 1-tert-butyl 2-methyl (21S)-4,4-difluoropyrrolidine-l,2-dicarboxylate. |
|
With (bis-(2-methoxyethyl)amino)sulfur trufluoride; In chloroform; at 0 - 20℃; for 19.0833h; |
Step 15-2: Synthesis of 1-tert-butyl 2-methyl (2S)-4,4-difluoropyrrolidine-1,2-dicarboxylate To a solution of 18.0 g of the compound obtained in Step 15-1 in CHCl3 (150 ml) was added dropwise 36.0 g of [bis(2-methoxy ethyl)amino]sulfur trifluoride under ice cooling over 5 minutes, then, the reaction mixture was stirred at room temperature for 19 hours. The reaction solution was added dropwise to a saturated aqueous solution of K2CO3 over 10 minutes under ice cooling. After liquid separation, the aqueous layer was extracted, the combined organic layer was washed with saturated brine, dried over MgSO4, then, the drying agent was separated by filtration, and the solvent was evaporated under reduced pressure. The obtained residue was purified by column chromatography (silicagel 60; mobile phase: EtOAc/n-hexane=1/4; v/v) to obtain 15.4 g of the title compound (yellow oil). MS (ESI pos.) m/z: 288 ([M+Na]+) 1H-NMR (300 MHz, CDCl3) delta (ppm); 1.43 & 1.46 (each-s, 9H), 2.36-2.56 (m, 1H), 2.58-2.82 (m, 1H), 3.69-3.92 (m, 2H), 3.77 (s, 3H) 4.40-4.61 (m, 1H) |
|
With diethylamino-sulfur trifluoride; In dichloromethane; at 0 - 20℃; for 11h; |
To a solution of 1-tert-butyl 2-methyl (2S)-4-oxo-1, 2-pyrrolidinedicarboxylate obtained in Example 8-1 (3.6g) in dichloromethane, was added diethylaminosulfur trifluoride (4mL) dropwise with cooling on an ice bath. The reaction mixture was warmed to room temperature and stirred for llhrs. To the resulting mixture was added NAHCO3, and the reaction mixture was quenched by adding saturated aqueous NAHCO3, and then water (1000mL) and CaCl2 (11.5g) in water (50mL) were added. The resulting suspension was extracted with ethyl acetate, the combined organic layer was washed with saturated aqueous NaCl, dried OVER MGS04, and concentrated in vacuo. The residue was purified with silica gel chromatography (n-hexane/ethyl acetate = 5/1) to give the target compound as a yellow oil (3.6g). H-NMR (INCDCL3) : D 1. 53-1.36 (9H, m), 2.55-2. 35 (1H, M), 2.83-2. 58 (1H, m), 3.94-3. 71 (5H, m), 4.62-4. 37 (1H, m). MS (ESI+) : m/z 266.18 (M+H). |
| 0.15 g |
With ethanol; diethylamino-sulfur trifluoride; In dichloromethane; at 20℃; for 18h;Inert atmosphere; |
Step 2: (S)-1-fert-butyl 2-methyl 4,4-difluoropyrrolidine-1 ,2-dicarboxylate A solution of the (S)-1 -ieri-butyl 2-methyl 4-oxopyrrolidine-1 ,2-dicarboxylate (0.23 g, 0.946 mmol), obtained from step 1 , in CH2CI2 (3.0 m l_), in a 25-mL flask equipped with a N2 inlet tube and stirring bar, was treated with a solution of diethylaminosulfur trifluoride (0.197 ml, 1 .607 mmol) in CH2CI2 (2.0 m l_) at room temperature. Ethanol (0.01 1 ml, 0.1 89 mmol) was added (for in situ generation of catalytic quantities of HF) and the mixture was stirred for 18h at room temperature. The solution was poured into saturated sodium bicarbonate and after C02 evolution ceased it was extracted into CH2CI2 (3 x 15 ml_), dried (Na2S04), filtered, and evaporated in vacuo. Chromatography on silica gel in DCM afforded a yellowish oil. Yield: 0.150g, 61 %. 1 H NMR (400 MHz, CDCI3): delta 1 .44 (br s, 9H), 2.45 (dq, J= 13.6, 5.2 Hz, 1 H), 2.61 -2.81 (m, 1 H), 3.75 (s, 3H), 3.60-3.90 (m , 2H), 4.45-4.55 (m, 1 H). MS (ESI) m/z 266.1 [M + H]+ |
|
With diethylamino-sulfur trifluoride; In dichloromethane; at -78 - 20℃; for 16.5h;Inert atmosphere; |
To a stirred solution of (S) -1-tert-butyl 2-methyl4-oxopyrrolidine-1, 2-dicarboxylate (5 g, 20.55 mmol) in dry DCM (100 mL) at-78 was added diethylaminosulfur trifluoride (DAST, 8.5 mL, 61.66 mol) dropwise over a period of 30 minutes under nitrogen atmosphere. The reaction mixture was stirred at this temperature for 1 h then at room temperature for 16 h. The reaction mixture was poured into the mixture of crushed ice and saturated NaHCO3aqueous solution till pH 8. The two layers were separated and the aqueous layer was extracted with DCM (20 mL×3) . The combined organic layer was washed with brine, dried over anhydrous Na2SO4then concentrated to get crude product (4.7 g, yellow oil) which was used in next step directly. MS: M/e 210 (M-55)+. |
|
With diethylamino-sulfur trifluoride; In ethanol; dichloromethane; at 0 - 20℃; for 18h; |
Step B: To a solution of N-Boc-4-oxo-L-methyl prolinate (106.00 g, 435.75 mmol) in dichloromethane (500 mL) was added DAST (119.40 g, 740.78 mmol) in dichloromethane (500 mL) dropwise at 0 C. After the addition, the mixture was added with EtOH (4.02 g, 87.15 mmol) and the mixture was stirred for 18 h at 0-20 C. The mixture was poured into icy sat.aq NaHCO3 slowly. After CO2 overflowed totally, the mixture was extracted with dichloromethane (2000 mL) twice, and the dichloromethane was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated in vacuo to give crude product. The crude product was purified by flash column chromatography eluted with Pet. Ether/EtOAc (50:1 to 10:1) to give N-Boc-4,4-fluor-L-methyl prolinate (70.00 g, 263.90 mmol, 60.56%). 1H NMR (CDCl3, 400 MHz): delta 4.57-4.40 (m, 1H), 3.89-3.72 (m, 5H), 2.78-2.60 (m, 1H), 2.45 (m, 1H), 1.44 (d, J = 20.0 Hz, 9H). |
|
With diethylamino-sulfur trifluoride; In dichloromethane; at 0 - 20℃; |
To a solution of 1-(tert-butyl) 2-methyl (S)-4-oxopyrrolidine-1,2-dicarboxylate (10 g, 41.1 mmol, 1.0 eq) in CH2Cl2 (80 mL) at 0 C. was added dropwise a solution of DAST (12.2 g, 75.7 mmol, 10.0 mL, 1.84 eq) in CH2Cl2 (20 mL). The resulting mixture was allowed to warm slowly to RT, stirred for a further 16 hr and quenched by dropwise addition of saturated aq. NaHCO3 (50 mL) and H2O (50 mL). The layers were separated, the aqueous phase was extracted with CH2Cl2 (50 mL×3) and the combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure to give the title compound as a viscous oil (11 g). 1H NMR (400 MHz, CDCl3): delta 4.55-4.43 (m, 1H), 3.89-3.81 (m, 2H), 3.77 (s, 3H), 2.71-2.65 (m, 1H), 2.52-2.44 (m, 1H), 1.47 (s, 9H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping