| 79% |
With sulfuric acid; dihydrogen peroxide; In methanol; at 20℃; for 0.5h;Inert atmosphere; |
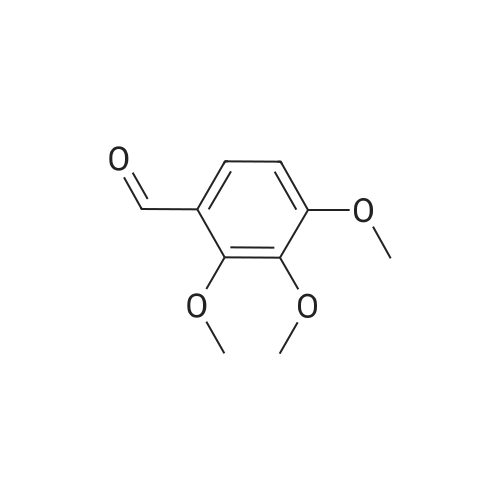
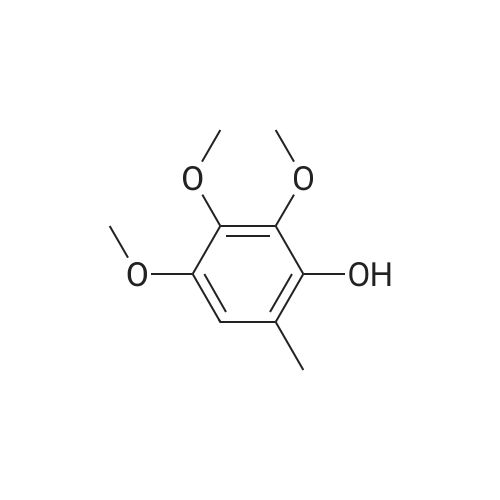
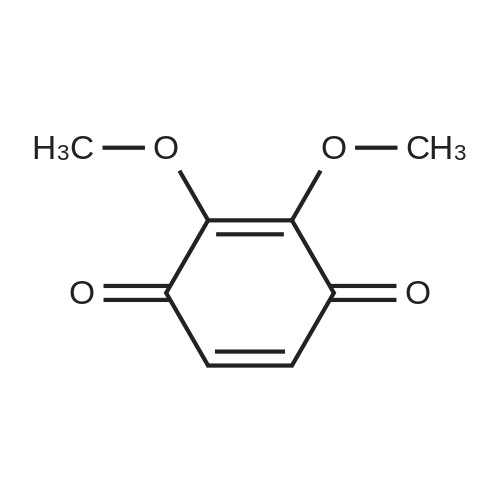
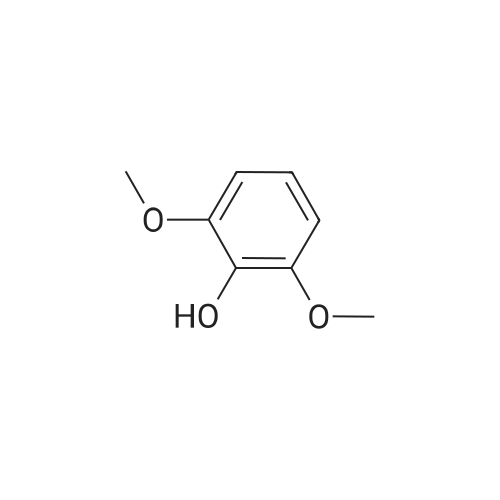
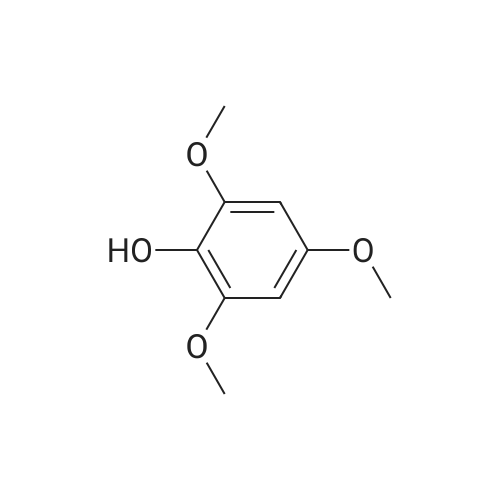
4.1.7 2,3,4-Trimethoxyphenol (15) H2O2 (30%, 13.6 mL, 131.2 mmol) was added dropwise at 0 C to a solution of 2,3,4-trimethoxybenzaldehyde (20 g, 100.9 mmol), and sulfuric acid (2 mL) in MeOH (200 mL) and the resulting mixture was stirred at room temperature for 30 min. The reaction was evaporated and extracted with EtOAc. The organic layer was collected and purified by column chromatography to afford 15 (14.8 g, 79%) as a transparent oil. 1H NMR (300 MHz, CDCl3): δ 3.79 (s, 3H), 3.88 (s, 3H), 3.94 (s, 3H), 5.54 (s, 1H), 6.54 (d, J = 9.0 Hz, 1H), 6.62 (d, J = 9.0 Hz, 1H). |
|
With sulfuric acid; dihydrogen peroxide; In methanol; at 25℃;Inert atmosphere; |
INTERMEDIATE 12,3,4-Trimethoxyphenol; A solution of 2,3,4-trimethoxybenzaldehyde (1,00 g, 5.10 mmol) and 30 wt/v % hydrogen peroxide (0.672 mL, 6.52 mmol) in cone. H2SO4 (0.102 mL) and MeOH (10.19 niL) was stirred overnight at 25 0C under N2. After this time the mixture was diluted with water (20 mL) and extracted with CH2Cl2 (3 x 30 mL). The combined extracts were dried (MgSO4) and concentrated in vacuo to afford the crude product. This was purified by flash chromatography (Biotage Horizon, 4OM, Si9 -30 niL/min, 100% hexanes for 360 mL, gradient to 50% EtOAc in hexanes over 2088 mL) to afford 2,3,4-trimethoxyphenol, as a colorless oil. R/ = 0.93 (50% EtOAc/hexanes). LCMS calc. = 185.1; found = 185.2 (M+H)+. 1H NMR (600 MHz, CDCl3): δ 6.62 (d, J- 9.0 Hz5 1 H); 6.55 (d, J= 8.9 Hz, 1 H); 5.49 (s, 1 H); 3.94 (s, 3 H); 3.89 (s, 3 H); 3.80 (s, 3 H). |
|
With sulfuric acid; dihydrogen peroxide; In methanol; at 20℃; for 12h; |
To a solution of 2,3,4-trimethoxybenzaldehyde (1.0 g, 5.1mmol) in MeOH (7.3 mL), 31% H2O2 (0.75 mg, 6.6 mmol)and H2SO4 (0.07 mL) were added at room temperature.After stirring for 12 h at the same temperature, the volatilesolvent was removed in vacuo to leave a residue. Theresidue was treated with H2O (30 mL) and extracted twicewith CHCl3. The extract was washed with H2O, saturatedNaHCO3 aqueous solution, and brine. The organic layerwas dried over Na2SO4 and concentrated in vacuo to givecrude 2,3,4-trimethoxyphenol (1.1 g), which was used forthe next step without further purification.1H NMR (CDCl3; 400 MHz) δ 6.63 (1H, d, J = 9.0 Hz),6.56 (1H, d, J = 9.0 Hz), 5.39 (1H, s), 3.96 (3H, s), 3.90(3H, s), 3.81 (3H, s). |
| 79 g |
|
To a solution containing 150.0 g ( 0.77 mol) of 2,3,4-trimethoxybenzaldehyde in 1000 mL of DCM was added 300.0 g (1.74 mol) of m- CPBA in five portions (30 g each) at 0C - 10C (ice-water bath). After the addition the reaction mixture was warmed to room temperature and stirred overnight. The reaction mixture was filtered to remove the solid and the filtrate was washed with aqueous NaHC03 (400 mL c 3), water (300 mL) and brine (300 mL). The organic layer was separated and dried over anhydrous Na2S04 and the mixture was filtered. The filtrate was concentrated to provide a dark yellow colored oil which was dissolved in EtOH (600 mL) and treated with a 10% aqueous KOH solution (500 mL) in one portion. The mixture was stirred at 50C for 4 h. The mixture was then cooled and acidified to pH=1 with 1 M HCI and extracted with DCM (500 mL x 3). The combined organic extracts were washed with water (500 mL) and brine (500 mL), dried over anhydrous Na2S04 and then filtered. The filtrate was concentrated and purified by silica gel chromatography (column height: 50 cm, diameter: 20 cm, 100-200 mesh silica gel, petroleum ether / EtOAc = 30/1 , 20/1 , 15/1 , 10/1) to give Int V-1 (79.0 g) as yellow oil. 1H NMR: (CDCIs, 400 MHz): d 6.63 (d, J = 8 Hz, 1 H), 6.55 (d, J = 8 Hz, 1 H), 5.38 (brs, 1 H), 3.96 (s, 3H), 3.90 (s, 3H), 3.81 (s, 3H). |
| 79.0 g |
|
Int V-1 To a solution containing 150.0 g (0.77 mol) of 2,3,4-trimethoxybenzaldehyde in 1000 mL of DCM was added 300.0 g (1.74 mol) of m- CPBA in five portions (30 g each) at 0C - 10C (ice-water bath). After the addition the reaction mixture was warmed to room temperature and stirred overnight. The reaction mixture was filtered to remove the solid and the filtrate was washed with aqueous NaHC03 (400 mL c 3), water (300 mL) and brine (300 mL). The organic layer was separated and dried over anhydrous Na2S04 and the mixture was filtered. The filtrate was concentrated to provide a dark yellow colored oil which was dissolved in EtOH (600 mL) and treated with a 10% aqueous KOH solution (500 mL) in one portion. The mixture was stirred at 50C for 4 h. The mixture was then cooled and acidified to pH=1 with 1 M HCI and extracted with DCM (500 mL x 3). The combined organic extracts were washed with water (500 mL) and brine (500 mL), dried over anhydrous Na2S04 and then filtered. The filtrate was concentrated and purified by silica gel chromatography (column height: 50 cm, diameter: 20 cm, 100-200 mesh silica gel, petroleum ether / EtOAc = 30/1, 20/1, 15/1, 10/1) to give Int V-1 (79.0 g) as yellow oil. 1H NMR: (CDCIs, 400 MHz): d 6.63 (d, J = 8 Hz, 1H), 6.55 (d, J = 8 Hz, 1H), 5.38 (brs, 1H), 3.96 (s, 3H), 3.90 (s, 3H), 3.81 (s, 3H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping