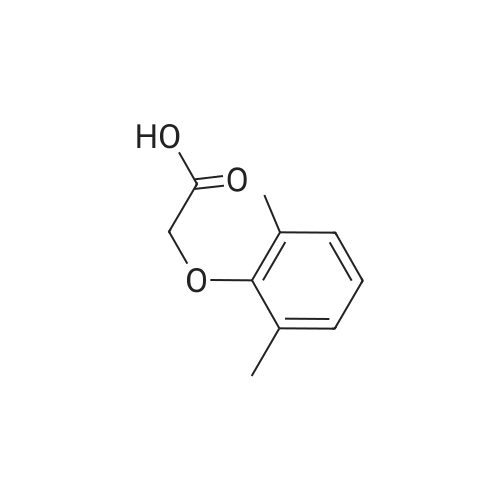
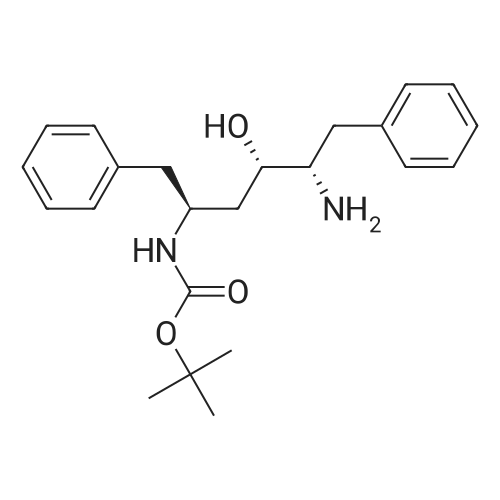
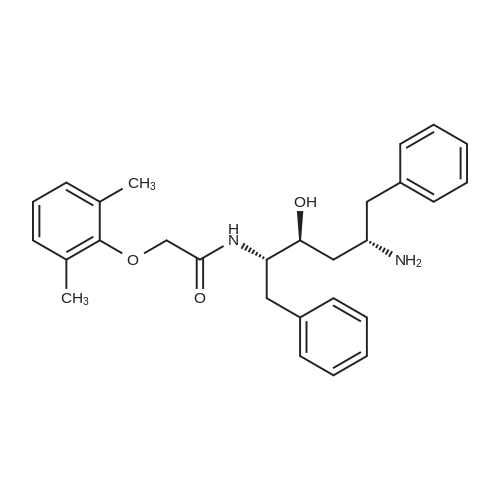
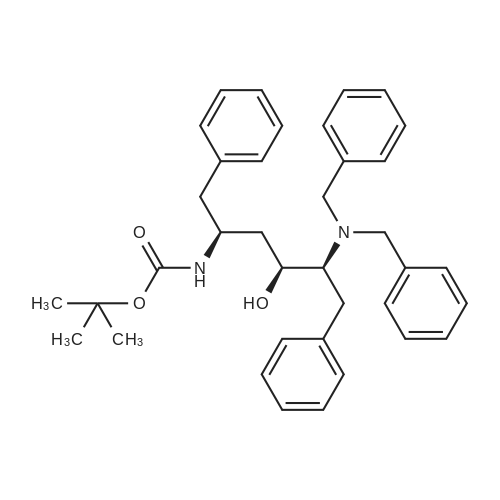
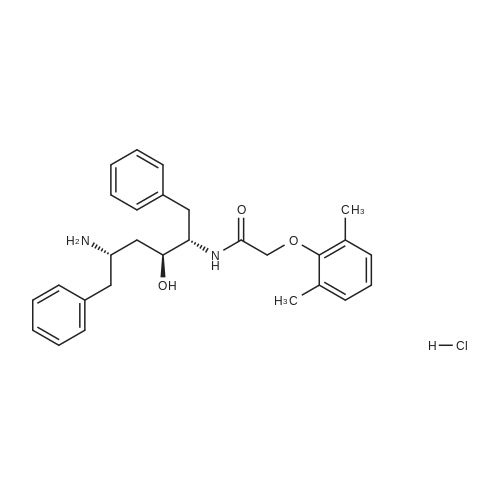
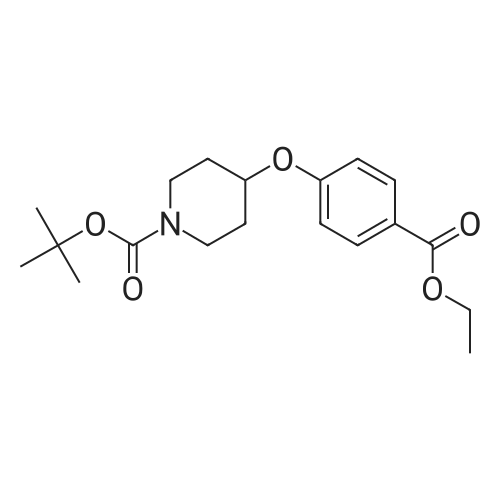
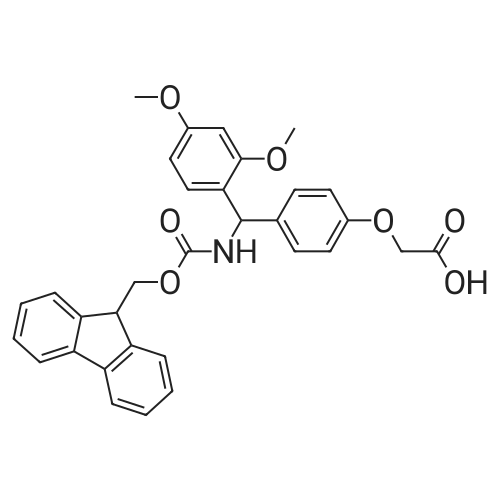
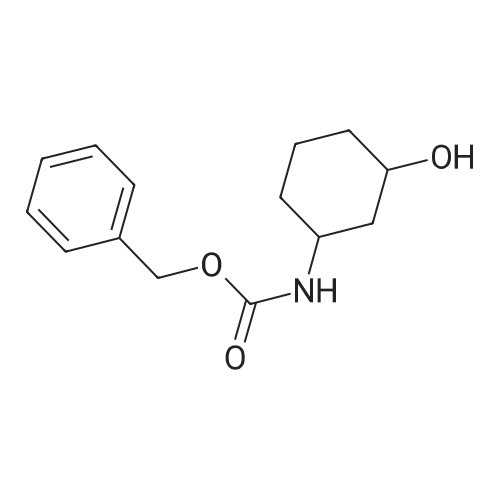
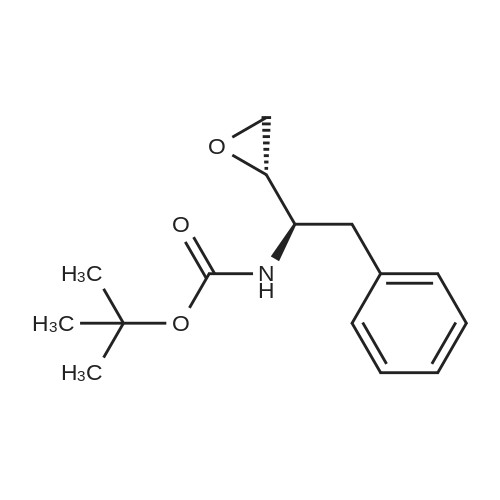
| 76.7% |
With 4-methyl-morpholine; benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 20℃; for 16h; |
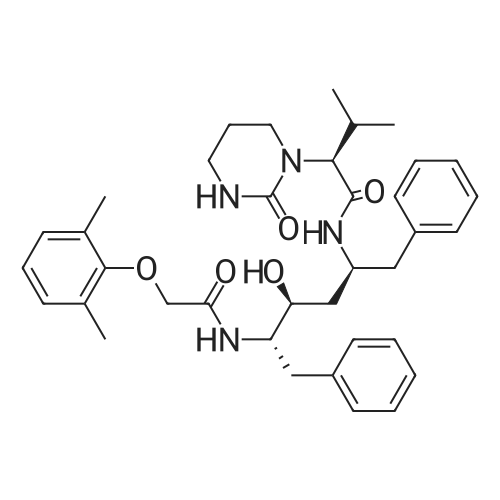
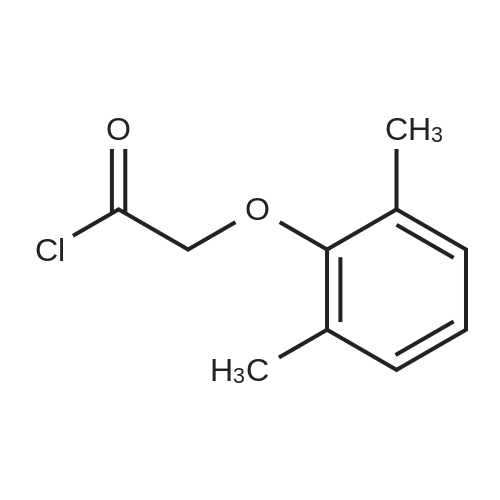
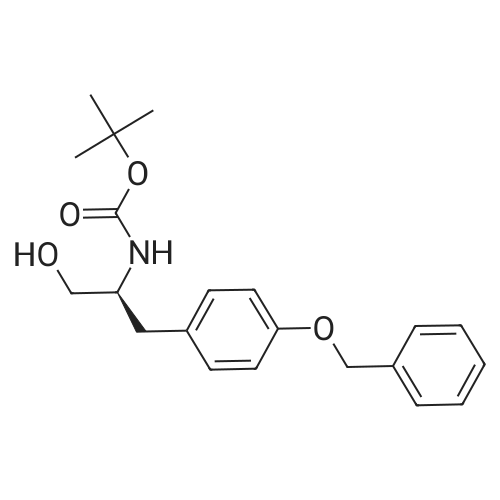
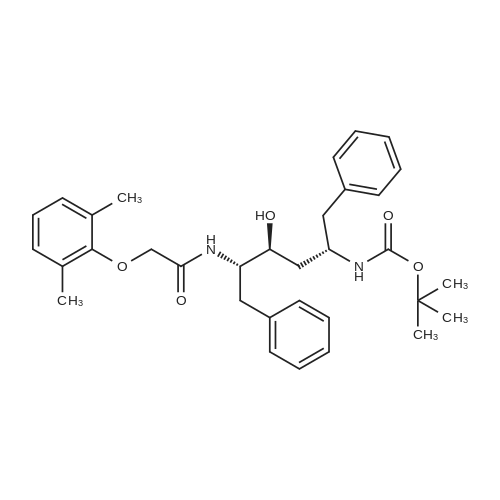
[00289] Example 18. Preparation of tert-butyl ( 1 S,3S,4S> 1 -benzyl-4- { [(2,6-dimethy.phenoxy)acetyl] amino} -S-hydroxy-5-phenylpentylcarbamate; [00290] A solution of /erf-butyl (lS,3S,4ιS)-l-benzyI-3-hydroxy-5-phenyl-4-amino-pentylcarbamate (38.5 mg, 0.1 mmol), (2,6-dimethyl-phenoxy)-acetic acid (18.9 mg, 1.05 equivalents), l-(3-dimethyl aminopropyl)-3-ethylcarbodiimide hydrochloride (29.7 mg, 1.5 equivalents), and 1-hydroxybenzotriazole (20.4 mg, 1.5 equivalents) in N,N-dimethylformamide (1 mL) was stirred for 4 minutes at room temperature. To this mixture was added N-methylmorpholine (27.5 μL, 2.5 equivalents) and the solution was stirred for 16 hours. The reaction mixture was quenched with saturated sodium bicarbonate, extracted with EtOAc, washed with 10% citric acid, dried (Na2SO4), and concentrated in vacuo. Column chromatography on silica (3% MeOH/ CH2CI2) gave a white solid (240 mg, 76.7%). IH NMR (300 MHz, DMSO-D6) δ ppm 1.31 (s, 9 H), 1.39 - 1.55 (m, J=6.99 Hz, 2 H), 2.14 (s, 6 H), 2.61 (d, ./=6.99 Hz, 2 H), 2.80 (d, 7=7.35 Hz, 2 H), 3.61 - 3.70 (m, 1 H), 3.84 (m, 1 H), 4.00 - 4.11 (m, 2 H), 4.20 - 4.38 (m, 1 H), 4.99 (d, 1 H), 6.66 (d, ./=9.19 Hz, 1 H), 6.88 - 7.28 (m, 13 H), 7.43 (d, 7=9.56 Hz, 1 H); MS m/z 547.4 (M+H)+. |
|
With O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate; triethylamine; In N,N-dimethyl-formamide; at 20℃; for 10h; |
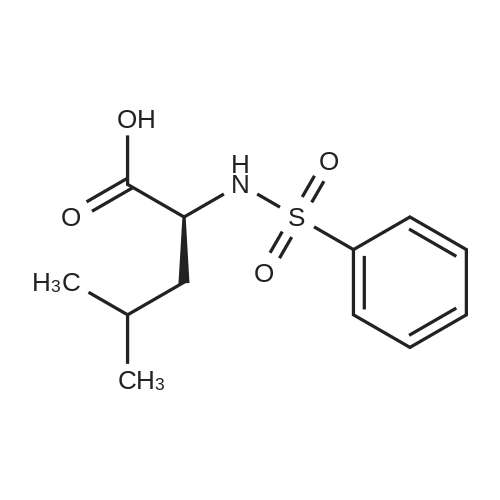
General procedure: To the mixture of compound 16 and the phenol relatedacetic acid derivative in DMF was added TBTU and Et3N,and stirred at room temperature for 10 hours. After evaporationof the solvent, the residue was purified on silica gel columnto afford 18, or 19, or 20 in 50-70% yield. By usingsimilar procedure, compounds 15 and 17 were converted into the amides 21 and 22. Products 18, 19, 20, 21 and 22 wereconfirmed by LCMS. Then compounds 21 or 22 were treatedby TFA/CH2Cl2 (9:1) for 30 min. After removal of the solventby co-evaporation with toluene, the residue was treatedwith N-phenylsulfonyl-valine in dry DMF in the presence ofTBTU and Et3N for 10 hours at room temperature under nitrogen.The volatiles of the reaction mixture were evaporatedand the residue was diluted with aqueous NaHCO3 and extractedwith CH2Cl2. Then the combined extracts wereevaporated and the residue was dissolved into ether and precipitatedwith hexane and filtered. The precipitated solidswere then dissolved into a mixture of CH2Cl2/MeOH, andmixed with a minimum amount of silica gel. After evaporatingthe volatiles, the silica gel adsorbed with the compoundswas loaded onto a silica gel column, and was washed withdichloromethane/ethyl acetate (3:1), then using methanol towash the column. The methanol fraction was concentrated,and the residue was suspended in hexane/ethyl acetate (1:1)and the solid product was filtered and washed with the samemixture to obtain the desired products 3, 4, 5, 23a, 23b and23c in more than 90% purities based on LCMS analysis. |
|
With 2-(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yl)-1,1,3,3-tetramethylisouronium hexafluorophosphate; triethylamine; In N,N-dimethyl-formamide; at 20℃; for 10h; |
(9) Add compound 17 and an equimolar amount of 2,6-dimethylphenol acetic acid to tetramethylurea tetrafluoroborate (TBTU), triethylamine (Et3N) and dimethylformamide (DMF) Stir at room temperature for 10 hours to obtain compound 22 respectively. Compound 22 was subjected to acidic conditions to remove the Boc group, 15 mg of compound 22 was dissolved in 9:1 trifluoroacetic acid: H2O, stirred at room temperature for 30 minutes, and toluene was added After evaporation to dryness, the obtained residue was coupled with N-benzenesulfonylvaline in a mixed solvent of tetramethylurea tetrafluoroborate, triethylamine and dimethylformamide to obtain compound 5, which was confirmed by LCMS The resulting compound 5 is the structure. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping