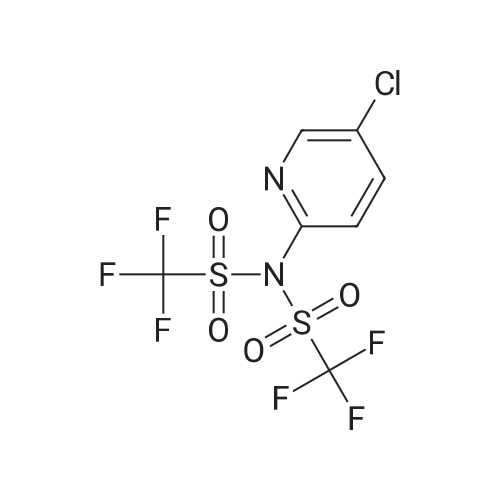
| 79% |
|
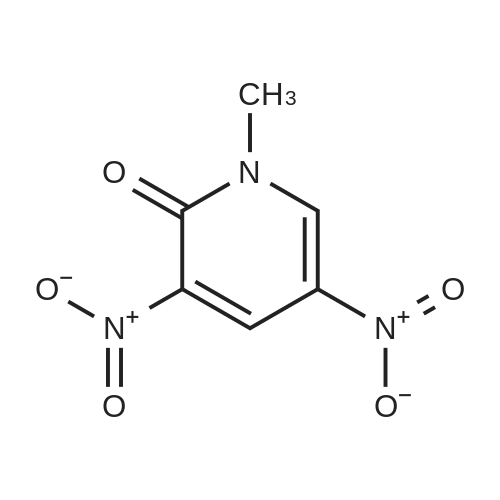
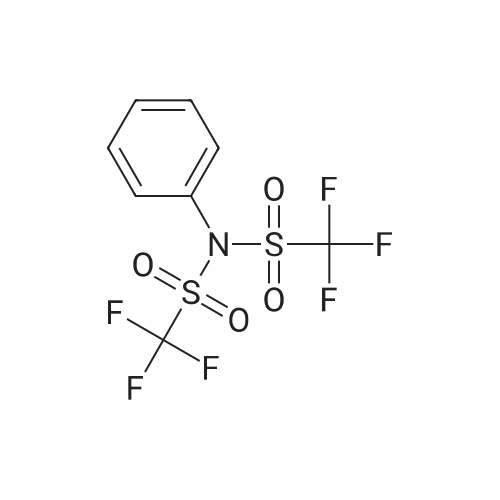
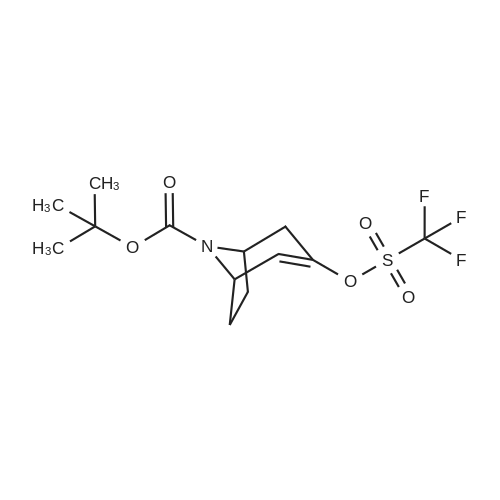
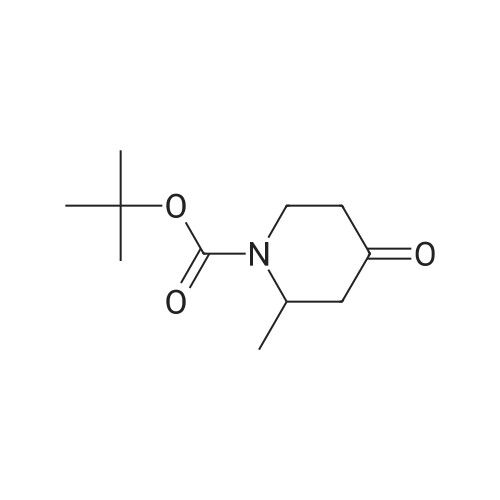
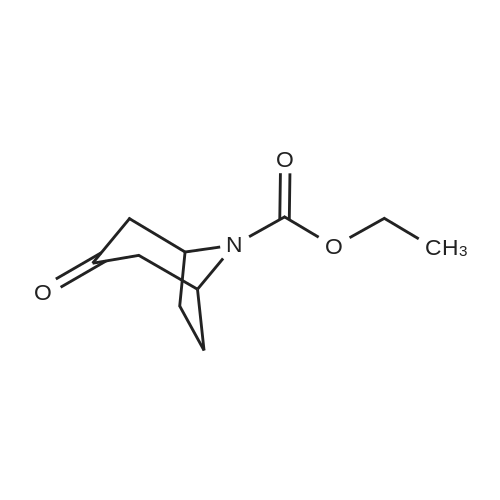
under nitrogen protection,3-Oxo-8-azabicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester (1.2 g, 5.3 mmol)The 15 mL THF solution was cooled to -70 C.LDA (1M in THF, 8 mL, 8.0 mmol) was added dropwise with stirring.The temperature of the control system is around -70 C.After stirring the system for 1 h, N-phenylbis(trifluoromethanesulfonyl)imide (1.9 g, 5.3 mmol) was added portionwise.After the addition, the system was naturally warmed under stirring and stirred at room temperature overnight.TLC showed that after completion of the reaction, the reaction system was poured into 100 mL of saturated ammonium chloride solution and extracted with EA (50 mL x 3).The organic phase is washed with saturated brine.Dry anhydrous Na2SO4 and concentrate under reduced pressure.The crude product was purified by column (PE:EA=20:1) to give 1.5 g.(yield: 79%) of the target compound,It is a white solid. |
| 72.6% |
|
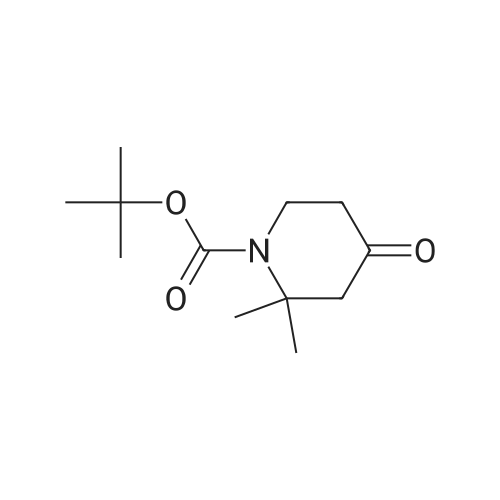
(0519) Tert-butyl 3-oxo-8-azabicyclo[3.2.1]octan-8-carboxylate (8.5 g, 37.8 mmol) was dissolved in tetrahydrofuran (80 mL), and a solution of lithium diisopropylamide in tetrahydrofuran/ n-heptane/ ethylbenzene (28 mL, 56 mmol, 2 M) was added slowly to the system at -78 C. After stirring for 10 min, a solution of 1,1,1-trifluoro-N-phenyl-N-((trifluoromethyl)sulfonyl)methanesulfonamide (14.8 g, 41.6 mmol) in tetrahydrofuran (50 mL) was added. After stirring for 30 min, the reaction was carried out at room temperature for 2 h. After the reaction, the mixture was concentrated to get the crude product. The crude product was purified by silica gel column chromatography (petroleum ether:ethyl acetate=20:1) to get the title compound (9.8 g, yield: 72.6%). |
| 63% |
|
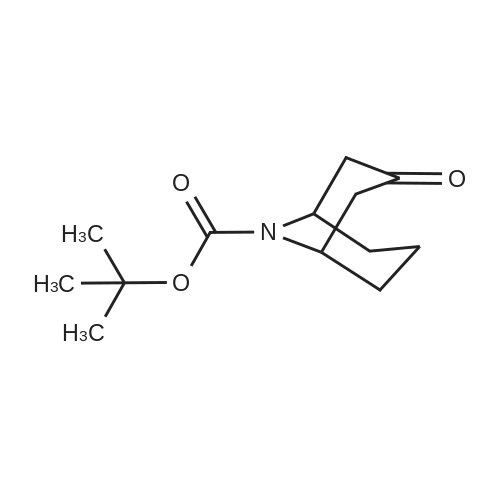
A. A 300 mL round bottom flask was charged with Compound 6a (1.0 g,4.44 mmol) and THF (30 mL). The mixture was cooled to -78 0C using a dry ice/acetone bath. A 20% solution of LiN(SiMe3)2 in THF (5.OmL, 5.32 mmol) was added dropwise over 15 min. The mixture was stirred at- 78 0C for 40 min. A solution of PhN(SO2CF3)2 (1.6 g, 4.48 mmol) in THF (33 mL) was added dropwise via addition funnel. The mixture was stirred for 18 h with gradual warming to room temperature. The mixture was concentrated in vacuo and purified via flash chromatography (230-400 mesh silica gel 60, 95:5 hexanes:EtOAc ) to give 1.O g (63%) of Compound 6b as a white solid. 1H NMR (300 MHz, CDCI3) delta 6.08 (d, J = 5.3 Hz, 1 H), 4.31-4.45 (m, 2 H), 3.04-3.21 (m, 2 H), 1.97-2.24 (m, 4 H), and 1.46 (s, 9 H). |
| 63.0% |
|
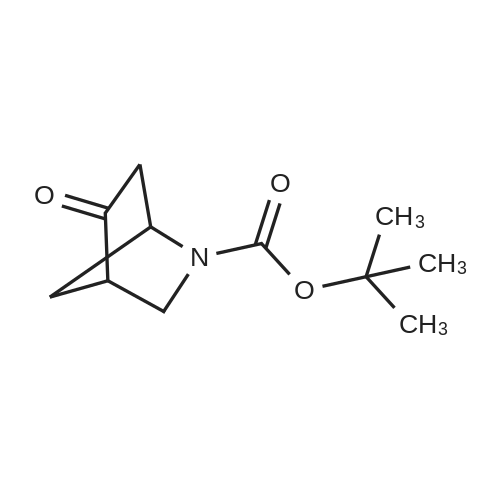
1360A. tert-Butyl 3-(((trifluoromethyl)sulfonyl)oxy)-8-azabicyclo[3.2.1]oct-3-ene-8-carboxylate To a stirred solution of tert-butyl 3-oxo-8-azabicyclo[3.2.1]octane-8-carboxylate (1.0 g, 4.44 mmol) in tetrahydrofuran (10 mL) at -78 C. was added LDA (3.33 mL, 6.66 mmol) and stirred at that temperature for 30 min. Then N,N-bis(trifluoromethylsulfonyl) aniline (1.586 g, 4.44 mmol) in tetrahydrofuran (5 mL) was added and stirred for 1 h. The reaction mixture was warmed to room temperature and stirred for 2 h. Reaction mixture was quenched with saturated ammonium chloride solution (10 mL) and extracted with ethyl acetate (2*100 mL). The organic layer was washed with brine solution (50 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure to get the crude, which was purified by silica gel flash chromatography to afford 1360A (brown oil, 1 g, 2.80 mmol, 63.0% yield). |
|
|
A solution of 3-oxo-8-azabicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester (15.8 g, 70.0 mmol) and THF (150 mL) was cooled to -78 C. and 1.0 M sodium hexamethyldisilazane in THF (84 mL) was added dropwise over 5 min. The reaction mixture was stirred for 1 h and then N-phenylbis(trifluoromethane-sulphonimide) (25.0 g, 70.0 mmol) was added and the reaction mixture was stirred for 1 h. The solution was warmed to room temperature, 1.0 N NaOH (100 mL) was added, and the reaction mixture was stirred for 15 min. Approximately 75 mL of solvent was evaporated. The resulting solution was diluted with ethyl acetate:hexanes (100 mL:100 mL) and water (100 mL), extracted and washed with 1.0 N NaOH (2*200 mL). The organic layer was washed with saturated NaCl solution (200 mL). The organic layer was collected, dried over anhydrous sodium sulfate, filtered, and concentrated to give the title compound (18.2 g) as a dark oil, which was used without further purification. |
|
With n-butyllithium; diisopropylamine; In tetrahydrofuran; hexane; n-heptane; at -60 - 20℃; for 24.25h; |
10 ml of a 2.5N solution n-butyl lithium in hexane are added, dropwise to a solution of 3.73 ml of diisopropylamine in 100 ml of tetrahydrofuran cooled to -60 C., in a 500 ml three-necked flask under nitrogen. After stirring for 1/4 hour, 5 g of N-tert-butyloxycarbonyl nortropinone in tetrahydrofuran (50 ml) at 0 C. are added. Finally, still at 0 C., 8.32 g of N-phenyltrifluoromethanesulfonimide are added. After stirring for 24 hours at ambient temperature, the tetrahydrofuran is evaporated off and the product is purified by rapid filtration over alumina, using a 2/1 mixture of heptane/ethyl acetate as eluent. 6.13 g of tert-butyl 3-[(trifluoromethyl)sulfonyl]oxy}-8-azabicyclo[3.2.1]oct-2-ene-8-carboxylate are obtained. |
|
|
1.1/3-Trifluoromethanesulfonyloxy-8-azabicyclo[3.2.1]oct-2-ene-8-carboxylic acid tert-butyl ester 3-Oxo-8-azabicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester (5 g, 22.2 mmol) is placed in 24 ml of anhydrous THF and the solution is cooled to -70 C. under nitrogen, 1N lithium bis(trimethylsilyl)amide in THF (24.4 ml, 24.4 mmol) is added dropwise. After stirring for 45 min at -70 C., N-phenyltrifluoromethane-sulfonimide (8.7 g, 24.4 mmol) placed in 25 ml of anhydrous THF is added dropwise. The temperature of the reaction medium is left to rise slowly. Stirring is maintained for 16 h at ambient temperature. After concentration to dryness, the crude product obtained is chromatographed on silica gel, elution being carried out with a cyclohexane/ether mixture (90/10). 10.2 g of expected 3-trifluoromethanesulfonyloxy-8-azabicyclo[3.2.1]oct-2-ene-8-carboxylic acid tert-butyl ester are obtained. [M+H+]=258 (-OtBu) |
|
|
To a solution of N-Boc-nortropinone (6 g, 26.6 mmol) in THF (70 ml) at -78C was added LDA (2 M in haptane/THF/ethyl benzene, 20ml, 40 mmol) slowly and the reaction mixture was stirred for 10 min. A solution of N~phenylbis(trifluoromethanesulfonimide) (10.5 g, 29.3 mmol) in THF (48 ml) was added. The reaction mixture was stirred at -78C for 30 min and the cooling bath was removed to warm it up to room temperature for 1.5 h until all N-Boc- nortropinone was utilized. Saturated NH4C1 solution (-10 mL) was added and stirring was continued for 5 minutes before the reaction mixture was transferred to a separatory funnel using EtOAc (150 mL). The organics were then extracted with EtOAc (2 x 125 ml), and washed with water (2 x 30 ml), brine (1 x 30 ml), and dried over MgS04 The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution withEtOAc/Hexanes (0-35%) gave the desired product, tert-butyl 3-(trifluoromethylsulfonyloxy)-8- azabicyclo[3.2. l]oct-3-ene-8-carboxylate. |
|
|
To a solution of tert-butyl 3-oxo-8-azabicyclo[3.2.1]octane-8-carboxylate (10 g, 44.4 mmol) in tetrahydrofuran (100 mL) was added 2M lithium diisopropylamide (26.6 mL, 53.3 mmol) dropwise at -60C under argon and the mixture was stirred at -60C for 1 hour. A solution of 1, 1,1-trifluoro-N- phenyl-N-((trifluoromethyl)sulfonyl)methanesulfonamide (17.44 g, 48.8 mmol) in tetrahydrofuran (100 mL) was added dropwise at -60C and the mixture was stirred at -60C for 30 minutes, and was allowed to warm to room temperature. The mixture was stirred under argon overnight, quenched with water (200 mL), and extracted with ethyl acetate (three times). The organic extracts were washed with 5% aqueous citric acid (twice) and stirred with 1M aqueous sodium hydroxide (200 mL) for 30 minutes. The wash process was repeated one additional time. The organic phase was dried over sodium sulfate, filtered, concentrated, and purified by flash chromatography on silica gel using an ISCO Companion eluting with ethyl acetate/petroleum ether (1 :20) to provide the title compound. |
|
|
To a 250 mL 3-necked round-bottom flask was placed a solution of (lR,5S)-tert- butyl 3-oxo-8-azabicyclo[3.2.1]octane-8-carboxylate (5.00 g, 22.2 mmol) in tetrahydrofuran (100 mL). Lithium bis(trimethylsilyl)amide (1.0 M in tetrahydrofuran, 26.6 mL, 26.6 mmol) was added dropwise at -78C. The mixture was stirred at -78C for 1 hour. Then a solution of l,l,l-trifluoro-N-phenyl-N-((trifluoromethyl)sulfonyl)methanesulfonamide (9.51 g, 26.6 mmol) in tetrahydrofuran (30 mL) was added at -78C. The mixture was stirred for an additional 16 h at ambient temperature. The solvent was removed in vacuo and the residue dissolved in ethyl acetate (100 mL), washed with water (2 x 50 mL) and brine (50 mL), dried over anhydroussodium sulfate and filtered. The filtrate was concentrated in vacuo and the residue purified on silica, eluting with ethyl acetate/petroleum ether (5: 100) to afford the title compound. LRMS (ESI) calc'd for Ci3Hi9F3N05S [M + H]+: 358, found 358; 1H NMR (400 MHz, CDC13) 56.08 (d, / = 5.4 Hz, 1H), 4.59-4.33 (m, 2H), 3.12-2.94 (m, 1H), 2.31-2.13 (m, 1H), 2.09-1.93 (m, 3H), 1.79-1.63 (m, 1H), 1.46 (s, 9H). |
|
|
Starting material 3-oxo-8-azabicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester (6.0 g, 26.63 mmol, 1.0 eq)Dissolved in anhydrous tetrahydrofuran solution (30 mL),Nitrogen protection drops to -70 C ~ -60 C,A solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran (1 mol/L, 32 mL, 1.2 eq) was added dropwise.After the drop, stir at -70 C ~ -60 C for 1 h,A solution of N-phenylbis(trifluoromethanesulfonyl)imide (11.4 g, 31.96 mmol, 1.2 eq) in tetrahydrofuran (30 mL).After the addition of 0 C or less, the reaction was carried out for 3 h, and the mixture was naturally stirred at room temperature for 12 h.The mixture was cooled to 0 C to 10 C, and brine (50 mL) and brine (50 mL)The filtrate was concentrated under reduced pressure to give a product. |
|
|
To a solution of ferf-butyl 3-oxo-8-azabicyclo[3.2.1]octane-8-carboxylate (Int 23a) (1.00 g, 4.33 mmol) in THF (10 mL) was added LDA (2 N, 3.3 mL) at -78 C and the mixture was stirred for 10 min at the same temperature. A solution of N- phenylbis(trifluoromethanesulfonimide) (1.75 g, 4.88 mmol) in THF (8 mL) was added. The mixture was stirred at -78 C for 30 min. The cooling bath was removed and the mixture was stirred for 1.5 h. Saturated aqueous NH4CI (30 mL) was added and stirring was continued for 5 min. The mixture was extracted with EtOAc (3 x 30 mL). The combined organic layers were washed with brine (30 mL), dried over anhydrous Na2S04, filtered and concentrated to dryness. The residue was purified by column chromatography on silica gel (EtOAc/PE = 1 :2) to give the title compound as a yellow oil. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping