| 93% |
With triethylamine; In dichloromethane; at 10℃; for 4h; |
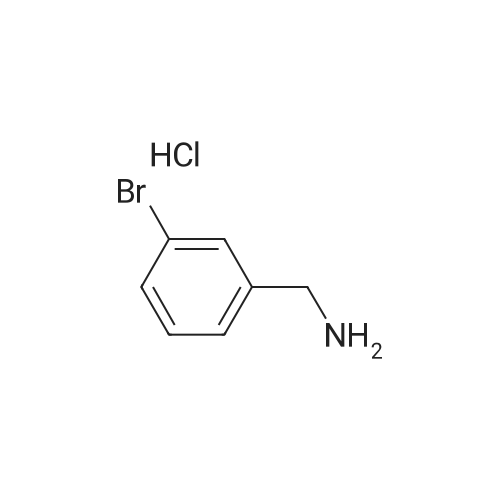
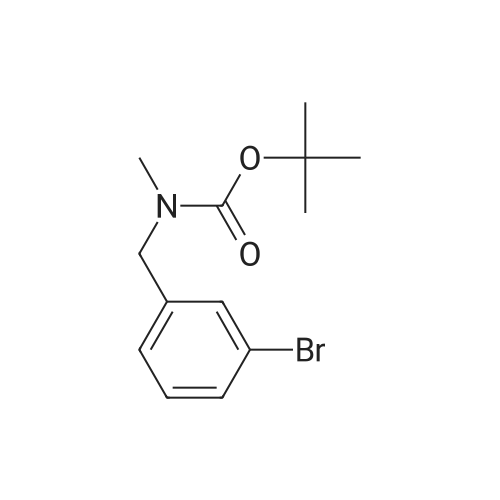
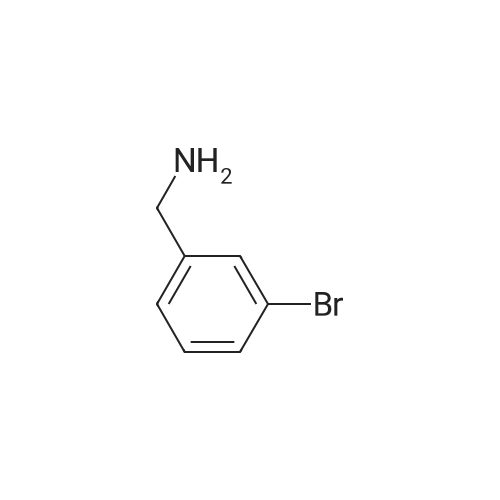
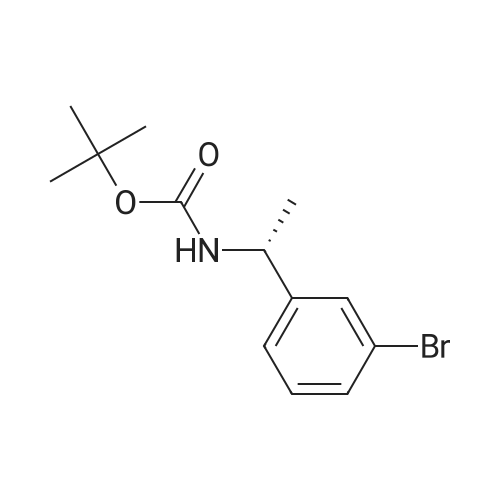
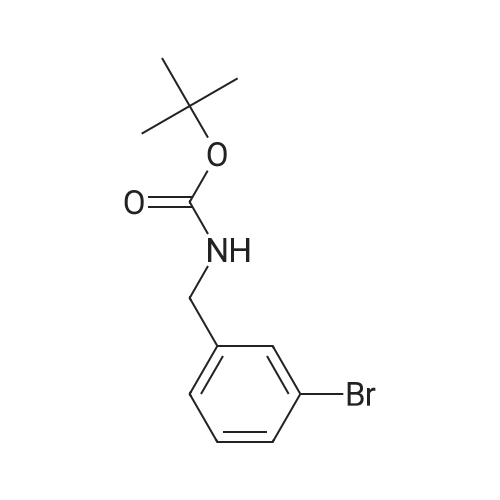
A solution of (3-bromophenyl)methanamine (1 g, 5.37 mmol) in dichloromethane (10 mL) at 10 C. was treated with triethylamine (1 .498 mL, 10.75 mmol) then fe/ -butyl dicarbonate (1 .498 mL, 6.45 mmol) and stirred at 10 C for 4 h. Water (10 mL) was added then the mixture extracted with DCM (2 x 10 mL), washed with brine, dried and evaporated and the residue purified by chromatography (silica gel, 10:1 hexanes/ethyl acetate) to afford the desired product (1 .5 g; 93%) as a colorless solid. NMR (500 MHz, CDCI3) delta ppm 7.43 (s, 1 H), 7.39 (d, J = 6.8 Hz, 1 H), 7.20 (d, J = 6.8 Hz, 2H), 4.89 (s, 1 H), 4.29 (d, J = 5.7 Hz, 2H), 1 .46 (s, 9H). |
| 92% |
With triethylamine; In dichloromethane; at 20℃; for 5.3h;Cooling with ice; |
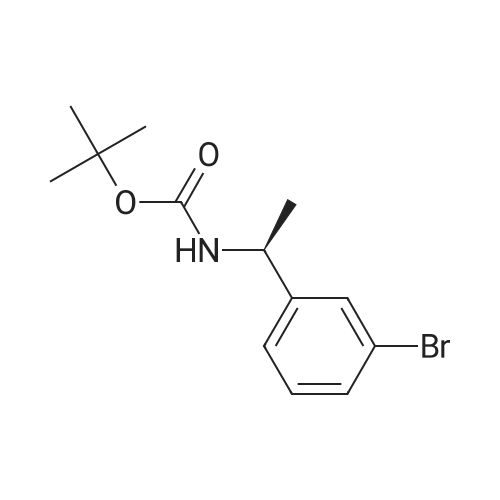
Example 104 Production of tert-Butly N-[(3-bromophenyl)methyl]carbamate. A methylene chloride (40 mL) solution of Boc2O(21.2 g, 97 mmol) was added dropwise to a methylene chloride (160 mL) solution of 3-bromobenzylamine (15.0 g, 81 mmol) and triethylamine (23 mL, 162 mmol) over 20 minutes, under ice-cooling, and the mixture was stirred at room temperature for 5 hours. The reaction mixture was washed with water, and an organic layer was then dried over anhydrous sodium sulfate. n-Hexane was added to the residue obtained by distilling off the solvent under a reduced pressure, and the precipitated solid was collected by filtration, to give the captioned compound (19.8 g, 92%). Mp 41-42C. 1H-NMR (DMSO-d6) delta: 1.39 (s, 9H), 4.12 (d, J=6.1 Hz, 2H), 7.23-7.28 (m, 2H), 7.40-7.42 (m, 3H). |
| 80% |
With triethylamine; In dichloromethane; at 20℃; |
To a stirred solution of benzylamine derivative (1 equiv.) in dry DCM, triethylamine (1 .5 equiv.) and di-tert-butyl dicarbonate (1 -1 .2 equiv.) were added under azote atmosphere. The reaction mixture was stirred at room temperature until completion. Water was then added, the phases were separated and the aqueous phase was extracted once with DCM. The combined organic layers were washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The crude mixture was then purified by flash column chromatography (0-10% MeOH in DCM) to provide the expected compound. The following compounds are examples illustrating this procedure:; tert-butyl bomophenyl)methyllcarbamate was prepared from 3- bromobenzylamine (2 g, 10.75 mmol). Yield: 2.47g (80%) of the title compound as a white powder |
|
With sodium hydroxide; In tetrahydrofuran; at 20℃; for 0.5h; |
A mixture of 3-aminomethylbromobenzene (22.11 g, 99.5 mmol), di-tert-butyl dicarbonate (26.07 g, 119.45 mmol), sodium hydroxide (8.76 g, 219 mmol) in tetrahydrofuran (75 mL) and water (100 mL) were stirred at room temperature for 30 minutes. Extraction and work-up with methylene chloride and water, followed by drying gave tert-butyl (3-bromo-benzyl) carbamate (34.9 g) |
|
|
Example 95 1-(3-Bromophenyl)-N-methylmethanamine To bromobenzylamine (0.890 g, 4 mmol) in THF (9 mL) was added NaOH (4.20 mL, 1 N, 4.20 mmol) and the solution was stirred at room temperature for 5 mins, when BOC2O (0.975 mL, 4.20 mmol) was added. This mixture was stirred for an additional 30 mins. The reaction mixture was diluted with EtOAc (20 mL). The organic layer was separated, washed with brine (5 mL), dried over Na2SO4, filtered and concentrated. Lithium aluminum hydride (12.00 mL, 12.00 mmol) was added to the above crude product and heated in a microwave at about 100 C. for about 1 h. The reaction mixture was diluted with Et2O (~50 mL) and quenched slowly with Na2SO4 (sat.). The organic layer was separated, dried over, filtered, and concentrated to afford the title compound (0.472 g, 59%). LC-MS m/z 200 (M+H)+. |
|
|
Example 167 1-(3-Bromophenyl)-N-methylmethanamine To bromobenzylamine (0.890 g, 4 mmol) in THF (9 mL) was added NaOH (4.20 mL, 1 N, 4.20 mmol) and the mixture was stirred at room temperature for 5 min where upon Boc2O (0.975 mL, 4.20 mmol) was added and that mixture was stirred for an additional 30 min. The reaction mixture was diluted with EtOAc (20 mL). The organic layer was separated, washed with brine (5 mL), dried over Na2SO4, filtered and concentrated. LAH (12.00 mL, 12.00 mmol) was added to the above crude product and heated in a microwave at 100 C. for 1 h. The reaction was quenched and diluted with Et2O (~50 mL) and quenched slowly with Na2SO4 (sat.). The organic layer was separated, dried over Na2SO4, filtered, and concentrated to afford the title compound (0.472 g, 59%). LC-MS m/z 200 (M+H)+. |
|
|
Example 95 l-(3-Bromophenyl)-lambda'-methylmethanamine To bromobenzylamine (0.890 g, 4 mmol) in THF (9 mL) was added NaOH (4.20 mL, 1 N, 4.20 mmol) and the solution was stirred at room temperature for 5 mins, when BOC2O (0.975 mL, 4.20 mmol) was added. This mixture was stirred for an additional 30 mins. The reaction mixture was diluted with EtOAc (20 mL). The organic layer was separated, washed with brine (5 mL), dried over Na2SOzI, filtered and concentrated. Lithium aluminum hydride (12.00 mL, 12.00 mmol) was added to the above crude product and heated in a microwave at about 100 0C for about 1 h. The reaction mixture was diluted with Et2O (-50 mL) and quenched slowly with Na2Stheta4 (sat.). The organic layer was separated, dried over, filtered, and concentrated to afford the title compound (0.472 g, 59%). LC-MS m/z 200 (M+H)+. |
|
With triethylamine; In dichloromethane; at 0 - 20℃; |
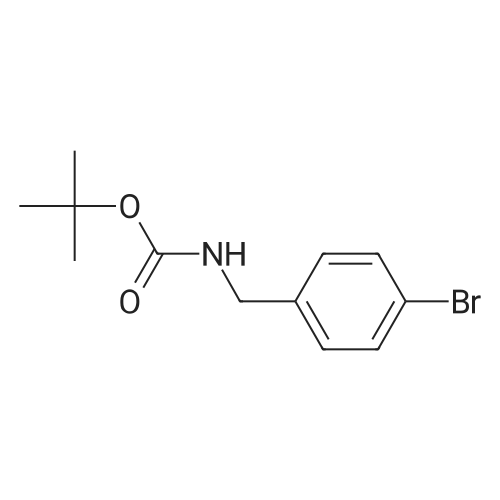
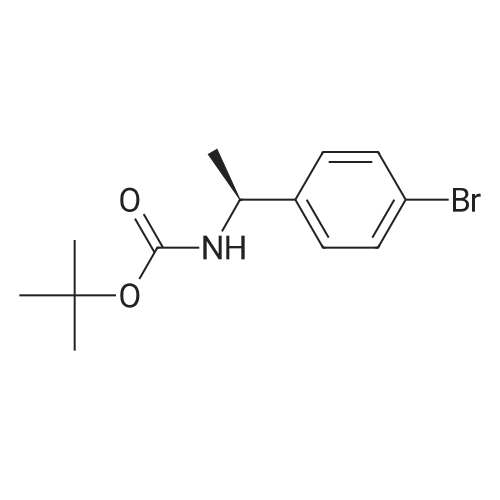
General procedure: (c) A solution of Boc2O (1.29g, 5.91mmol) in CH2Cl2 (10ml) was added to a suspension of 4-bromobenzylamine (1.00g, 5.38mmol) and Et3N (0.60g, 5.91mmol) in CH2Cl2 (10ml) for 5min at 0C with a CaCl2 tube. The reaction mixture was stirred overnight at room temperature. Then, H2O (50ml) was added to the mixture, and the organic layer was separated. The aqueous layer was extracted with CHCl3 (50ml×3). The combined organic layer was washed with H2O (50ml×1) and then brine (50ml×1), dried over Na2SO4 (anhyd), filtered, and concentrated under reduced pressure to afford (4-bromobenzyl)carbamic acid tert-butyl ester (1.63g, quant. y.) as a colorless solid. |
|
With sodium hydrogencarbonate; In tetrahydrofuran; at 20℃; |
Add m-bromobenzylamine (1.86 g, 10 mmol) to the round bottom flask, dissolve in 100 mL of THF, add sodium bicarbonate (1.68 g, 20 mmol), and add di-tert-butyl dicarbonate (2.4 g, 11 mmol) with stirring,Stir overnight at room temperature, spin off the solution, dissolve the EA, wash with saturated brine three times, dry the organic phase over anhydrous sodium sulfate, spin dry to obtain a white solid, and directly send to the next step |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping