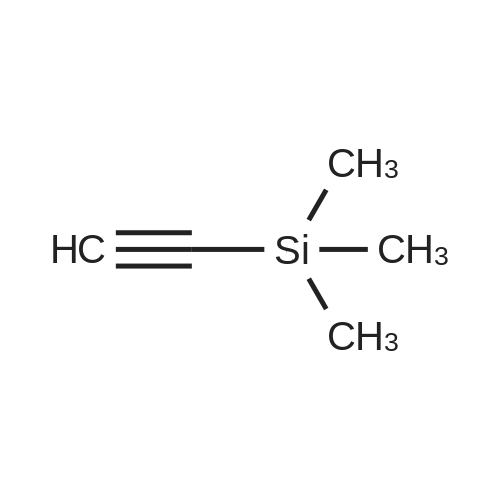
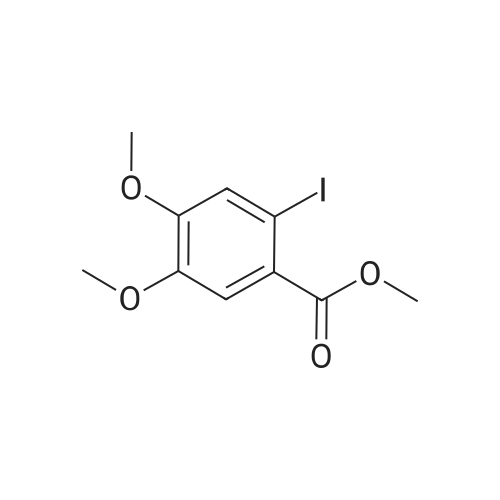
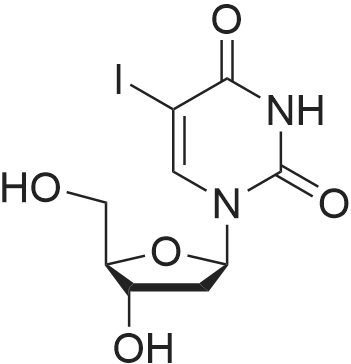
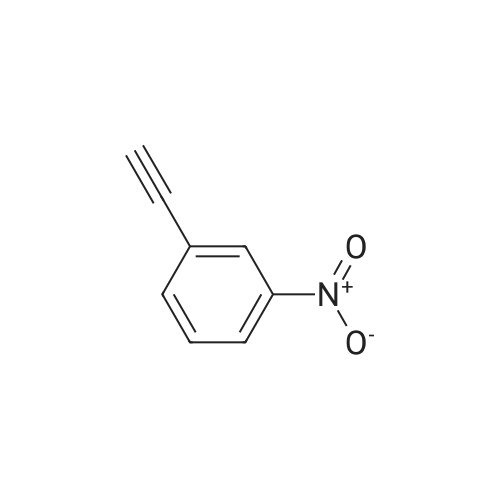
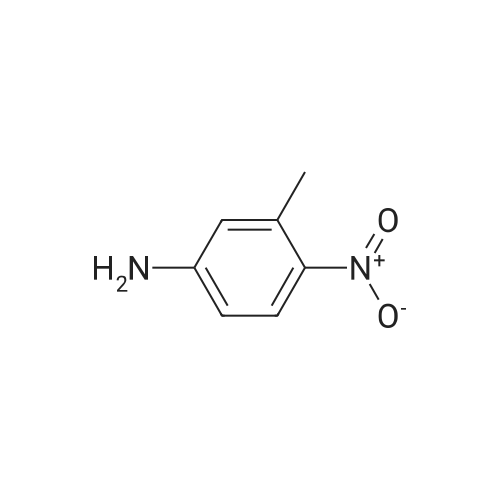
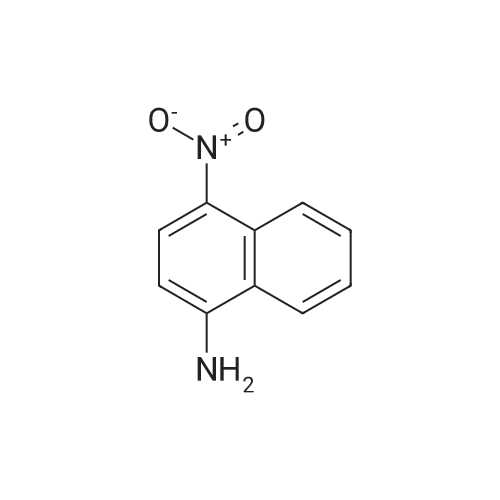
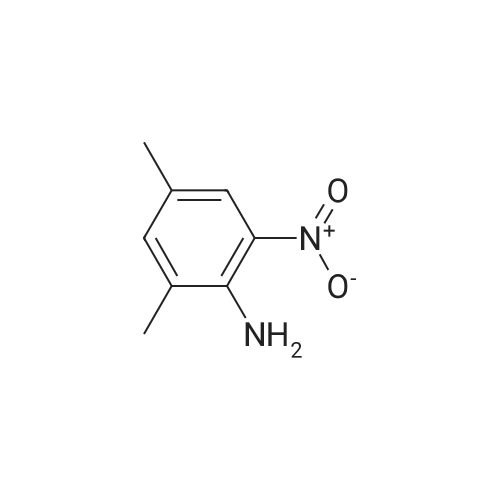
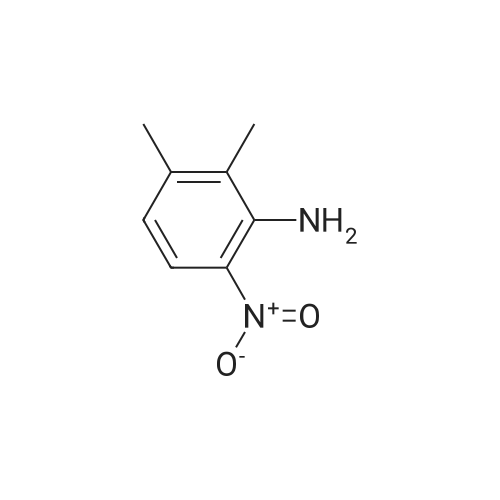
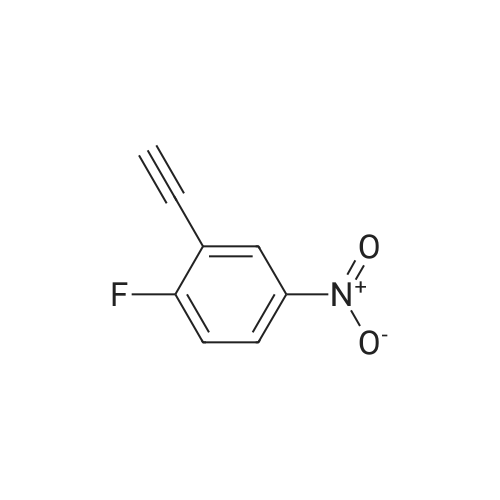
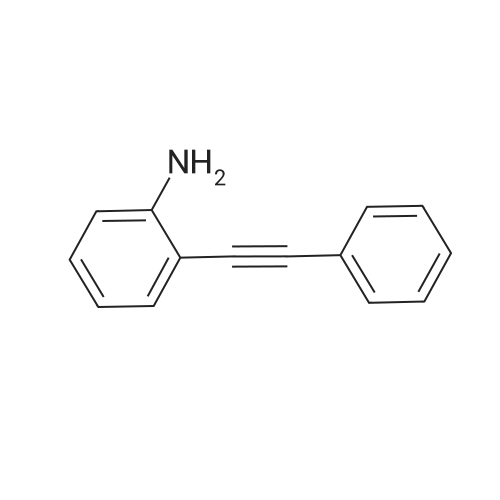
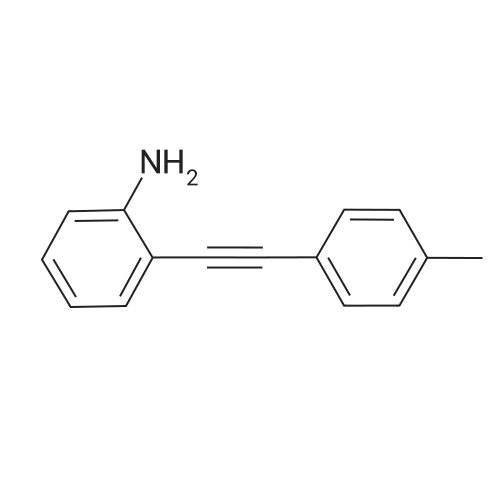
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With triethylamine; In chloroform; at 50℃; for 1.75h;
General procedure: A solution of the oxime (3.10 mmol) and N-chlorosuccinimide (0.414 g, 3.10 mmol) im 4 mL anhydrous chloroform was treated with pyridine (0.014 mL, 0.17 mmol) and heated under nitrogen at 50 C for 1 h. Then acetylene 5 (0.400 g, 2.72 mmol) was added, followed by dropwise addition of triethylamine (0.52 mL, 3.8 mmol) over 15 min. The mixture was heated at 50 C for another 90 min, then cooled, treated with 60 mL water and extracted with dichloromethane (3 x 20 mL). The combined organic layers were dried over sodium sulfate and evaporated to dryness. The residue was purified by flash column chromatography using hexanes-ethyl acetate mixtures.
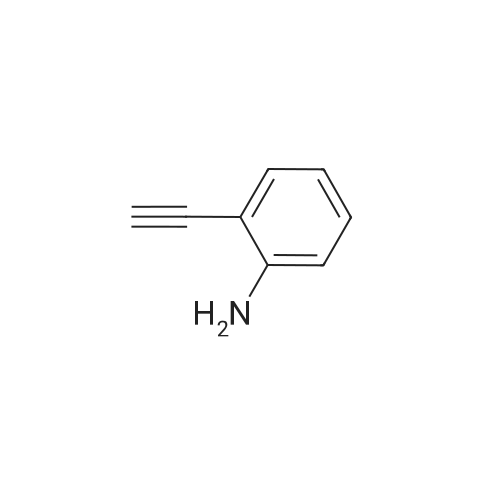
With palladium 10% on activated carbon; hydrazine hydrate; In ethanol;Heating;
To a solution of compound 6k (1.5 g, 10.19 mmol) in ethanol(20 mL), 80% hydrazine monohydrate (1.8 g, 20.39 mmol) and 10%Pd/C (20 mg) were successively added. The reaction mixture was stirred under reflux at 80 C for 2-3 h and the reaction was monitored by TLC. After completion of the reaction, the solution was filtered to remove the Pd/C and the solvent was removed under vacuum. Then water and ethyl acetate were added to the residue and the organic layer was separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water and brine, and dried over anhydrous sodium sulfate. The solvent was removed in vacuo to give pure 2-ethynylaniline as light yellow oil liquid, yield: 87.45%. 3-ethynylaniline (7l) and 4-ethynylaniline (7m) were prepared withthe same method and the corresponding compounds 6l and 6m.
With potassium carbonate; In methanol; at 20℃; for 0.166667h;
38 g (173.27 mmol) of Intermediate E-1 was stirred with500 ml of methanol in a 1000 mE flask. 24 g (173.27 mmol) of potassium carbonate was added thereto, and the mixture was stirred for 10 minutes at room temperature to complete a reaction. The reactant was filtered, water and ethylacetatewere respectively added thereto in an amount of 500 ml after removing the potassium carbonate, and the water was separated through an extraction. The separated organic solvent was removed through a distiller to obtain Intermediate E-2(25.1 g, 98%).calcd. C8H5N02: C, 65.31; H, 3.43; N, 9.52; 0, 21.75; found: C, 65.25; H, 3.47; N, 9.56; 0, 21.58.
66%
With tetrabutyl ammonium fluoride; In tetrahydrofuran; at -10 - 20℃; for 2h;Inert atmosphere;
To a 3-neck RBF (2000 ml_) was added compound B (108g, 0.492mol), followed by the addition of THF(500 ml_). The mixture was cooled to -10 C under N2. The Bu4NF in THF was added dropwise below 0 C. After addition the reaction mixture was warmed to ambient temperature and stirred for two hours. TLC checked the reaction was completed. The reaction mixture was poured into water while keeping the temperature below 30 C, extracted with MTBE, washed with water, dried with sulfate sodium. After filtration the filtrate was concentrated on vacuum to give the crude product. Purification by Silica gel column chromatography gave the product (48g, yield: 66%).
62%
With tetrabutyl ammonium fluoride; In tetrahydrofuran; at 20℃; for 1.5h;Inert atmosphere;
Preparation of Compound 1-2 [105] After dissolving Compound 1-1 (39g, 178mmol) in THF (1.2L) at nitrogen atmosphere, tetrabutylammonium fluoride (280mL, 1M solution in THF, 213mmol) was added thereto. The reaction mixture was stirred at room temperature for 1.5 hours and extracted with distilled water and ethylacetate. After drying an organic layer with anhydrous MgSO4 and removing a solvent by a rotary type evaporator, Compound 1-2 (16.2g, 62%) was obtained via purification by column chromatography using methylene chloride (MC) and hexane as a developing solvent.
With methanol; potassium carbonate; In dichloromethane; at 20℃; for 5h;
8.022 g (36.6 mmol) of Intermediate (2) and 5.56 g (40.2 mmol) of K2CO3 were added to 20 mL of methanol and 80 mL of methylene chloride (DCM), and the resultant mixture was stirred at ambient temperature. After 5 hours, the completed reaction mixture was added to 300 mL of distilled H2O and an organic layer was extracted therefrom by using DCM. The extracted organic layer was dried by using MgSO4 and a solvent was removed therefrom by using an evaporator. A concentrated product was purified therefrom by silica gel column chromatography to obtain Intermediate (3).
2.07 g
With methanol; potassium carbonate; at 20℃; for 0.166667h;
General procedure: According to the reported method, 1 the title compound 1b was prepared from 1-bromo-4-chlorobenzene by the Sonogashira coupling and subsequent desilylation as follows. Under an argon atmosphere, TMSA (2.58 g, 26.2 mmol) was added to a stirred mixture of 1-bromo-4-chlorobenzene (3.83 g, 20.0 mmol), PdCl2(PPh3)2 (350 mg, 0.499 mmol) and CuI (95.2 mg, 0.500 mmol) in Et3N (40 mL). The mixture was heated to 80 C and stirred for 19 h. The resultant mixture was cooled to room temperature and passed through a short silica gel-activated carbon column. Evaporation of the filtrate under reduced pressure gave a light yellow solid (4.40 g). The crude product was dissolved in MeOH (40 mL) and treated with anhydrous K2CO3 (4.37 g, 31.6 mmol) at room temperature for 15h. The reaction mixture was quenched by dropwise addition of HCl (2 M in H2O, 10 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic layer was washed with brine, dried overNa2SO4, and evaporated under reduced pressure. Purification of the residue by silica gel column chromatography (hexane) gave 1b as pale yellow solid (1.23 g, 9.22 mmol, 46%).
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In ethanol; for 8h;Inert atmosphere; Reflux;
To a RBF (1000 ml) was added compound C (25g, 0.17gmol), followed by the addition of Et3N (600 ml). The mixture was purged with N2 for 10 min. A catalyst amount of Pd (PPh3)2CI2 (200mg) and Cul (35mg) were added. The mixture was stirred for 6 hours, filtered and the solid was collected. The solid was reflux in ethanol (500ml_) for 2 hours and the solid was collected to give the crude product. The material was purified with crystallization from toluene to give the product (42g, 92%).
89%
With triethylamine;bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; at 20℃;
Preparation of Compound 1-3 [107] After 2-iodonitrobenzene (22.4g, 130mmol), triethylamine (1.5L), Pd(PPh3)2Cl2 (3.7g, 5.4mmol), CuI (1g, 2.2mmol) and Compound 1-2 (16g, 109mmol) were mixed, the mixture was stirred at room temperature overnight. After dissolving suspension, which is obtained after filtration, in MC and performing silica filtering, Compound 1-3 (26g, 89%) was obtained via recrystallization with ethanol.
38%
With pyrrolidine; copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); water; at 80℃; for 8h;
General procedure: To a stirred mixture of 2-iodonitrobenzene (249 mg, 1.0 mmol), phenyl acetylene(153 mg, 1.5 mmol), pyrrolidine (142 mg, 2 mmol), Pd(PPh3)4 (12 mg,0.01 equiv) and CuI (2 mg, 1 mol %) were added followed by water (5 mL).The reaction mixture was then stirred at 80 C (oil bath temperature) for 8 h(TLC) and extracted with ethyl acetate (3 20 mL). The combined organicextract was washed with brine, dried over anhydrous Na2SO4, and evaporated to leave the crude product which was purified by column chromatography oversilica gel with hexane-ethyl acetate (90:10) as eluent to furnish pure 2-(2-nitrophenyl)-1-phenylethanone
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In tetrahydrofuran; at 50℃; for 3h;Inert atmosphere;
7.76 g (34.3 mmol) of 1-iodo-2-nitrobenzene (S), 5.05 g (34.3 mmol) of Intermediate (3), 1.20 g (1.72 mmol) of bis(triphenylphosphine)palladium(II)dichloride, and 0.65 g (3.43 mmol) of copper(I)iodide were dissolved in 60 mL of THF under an argon atmosphere. 10 mL of triethylamine was added thereto. Then, the resultant mixture was reacted at a temperature of 50 C. After 3 hours, a solvent was removed therefrom by using an evaporator. A concentrated product was purified therefrom by silica gel column chromatography to obtain Intermediate (4).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping