|
With carbon tetrabromide; triphenylphosphine; In dichloromethane; at 0℃; |
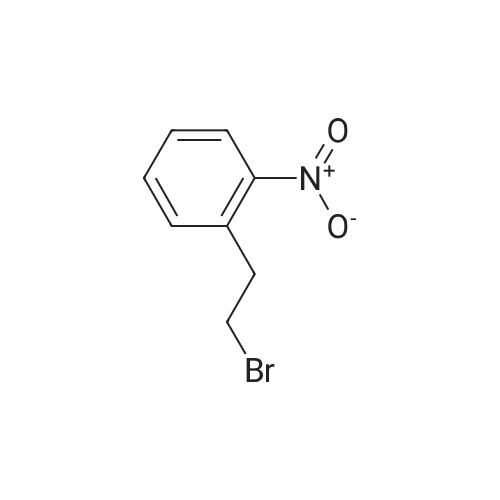
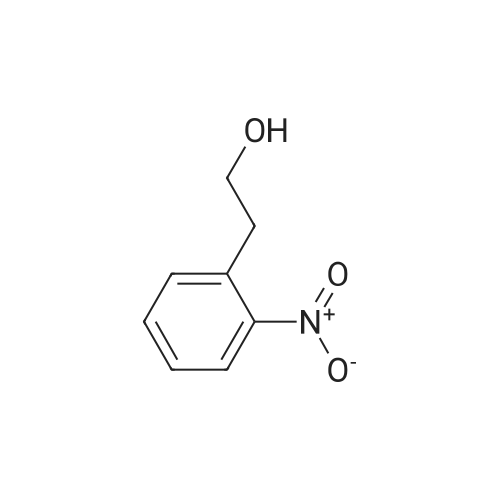
Triphenylphosphine (39.2 g, 0.150 mol) and carbon tetrabromide (49.5 g, 0.150 mol) were added sequentially to a solution of 2-(2-hydroxyethyl)-nitrobenzene (25.0 g, 0.150 mol) in methylene chloride (400 mL) at 0OC. The reaction was stirred overnight and quenched with saturated sodium bicarbonate solution. The methylene chloride phase was washed with saturated brine and dried over magnesium sulfate. The crude product was treated with ethyl acetate, and the precipitated triphenylphosphine oxide removed by filtration. Further purification by flash chromatography by (silica gel, 0-10percent ethyl acetate in hexane gradient elution) produced the title compound (27.9 g). |
|
With carbon tetrabromide; triphenylphosphine; In dichloromethane; at 0 - 20℃; |
1-(2-Bromoethyl)-2-nitrobenzene; To a solution of 1-(2-hydroxyethyl)-2-nitrobenzene (21 ml, 150 mmol) and triphenylphosphine (39.2 g, 150 mmol) in DCM (400 ml) at 0° C. was add CBr4 (49.5 g, 150 mmol) in portions and the reaction mixture was stirred from 0° C. to RT overnight. The reaction mixture was quenched with sat. aq. Na2CO3, the layers were separated and the organic layer was washed with brine, dried (MgSO4) and evaporated to dryness. The residue was treated with EtOAc and the precipitated Ph3O was filtered and the solvent removed. This was repeated twice more. Purification by column chromatography (0percent to 10percent EtOAc in Hx) gave an oil that solidified on standing. |
|
With carbon tetrabromide; triphenylphosphine; In dichloromethane; |
INTERMEDIATE 7 3 -f 4-PiperidinyD- 1 ,3 A5-tetrahydro-2H- 1 ,3 -benzodiazapin-2-one hydrochloride Step A. 2-(2-Bromoetfayl)nitrobenzene; Triphenylphosphine (39.2 g, 0.150 mol) and carbon tetrabromide (49.5 g, 0.150 mol) were added sequentially to a solution of 2-(2-hydroxyethyl)-nitrobenzene (25.0 g, 0.150 mol) in methylene chloride (400 mL) at O0C. The reaction was stirred overnight and quenched EPO <DP n="71"/>with saturated sodium bicarbonate solution. The methylene chloride phase was washed with saturated brine and dried over magnesium sulfate. The crude product was treated with ethyl acetate, and the precipitated triphenylphosphine oxide removed by filtration. Further purification by flash chromatography by (silica gel, 0-10percent ethyl acetate in hexane gradient elution) produced the title compound (27.9 g). |
|
With carbon tetrabromide; triphenylphosphine; In dichloromethane; at 0℃; |
Triphenylphosphine (39.2 g, 0.150 mol) and carbon tetrabromide (49.5 g, 0.150 mol) were added sequentially to a solution of 2-(2-hydroxyethyl)-nitrobenzene (25.0 g, 0.150 mol) in methylene chloride (400 mL) at O0C. The reaction was stirred overnight and quenched with saturated sodium bicarbonate solution. The methylene chloride phase was washed with saturated brine and dried over magnesium sulfate. The crude product was treated with ethyl acetate, and the precipitated triphenylphosphine oxide removed by filtration. Further purification by flash chromatography by (silica gel, 0-10percent ethyl acetate in hexane gradient elution) produced the title compound (27.9 g). |
|
With carbon tetrabromide; triphenylphosphine; In dichloromethane; at 0℃; |
(i) Triphenylphosphine (2.62 g, 10.0 mmol) and carbon tetrabromide (3.32 g, 10.0 mmol) were added sequentially to a solution of 2-(2-hydroxyethyl)-nitrobenzene(1.41 ml, 10.0 mmol) in dichloromethane (50 ml) at O0C. The reaction was stirred overnight and quenched with saturated aqueous sodium bicarbonate. The organic layer was washed with brine, dried over anhydrous magnesium sulphate and concentrated in vacuo. The residue was treated with ethyl acetate, and the precipitated triphenylphosphine oxide removed by filtration. Purified by flash-silica gel column chromatography, eluting with a 0-100percent gradient of ethyl acetate in hexane to yield 2-(2-Bromoethyl)-nitrobenzene (2.30 g). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping