* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
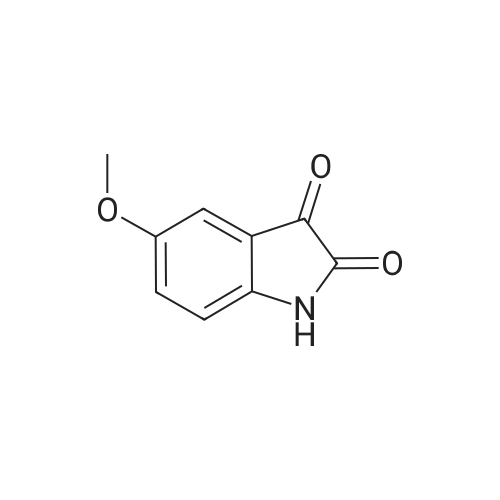
General procedure: Concentrated sulfuric acid (95 mL) was heated in a flask at 60 C. Intermediates 2 (12 g, 52mmol) were slowly added in portions to maintain the temperature below 70 C. The temperature was then increased to 80 C. After 3 h, the solution was cooled to room temperature, and then poured into crushed ice with vigorous stirring. The precipitate was collected by filtration and dried under vacuum to provide the product 3.
70%
at 55 - 80℃; for 0.166667 h;
The procedure described by Marvel et al. (see Org. Synth. Coll. Vol. I, 327) was followed. Intermediate 12 (11.9 g, 48.5 mmol) was added in small portions to 35 mL of concentrated sulfuric acid at 55° C. in a 250 mL Erlenmeyer flask. The temperature of the solution was maintained below 70° C. until all the acetamide had been added and it was increased to 80° C. for 10 minutes. The dark-colored solution was cooled to room temperature and poured onto 175 mL of crushed ice. After standing for 30 minutes, the precipitate was collected by filtration, washed three times with water, and dried under vacuum to yield indoline-2,3-dione of sufficient purity to be used in the next step (intermediate 13, 8.32 g, 70percent yield). 1H NMR (400 MHz, DMSO-d6) δ 7.15 (t, J=7.8 Hz, 1H), 7.56 (d, J=7.3 Hz, 1H), 7.64 (d, J=8.3 Hz, 1H), 11.71 (s, 1H).
59%
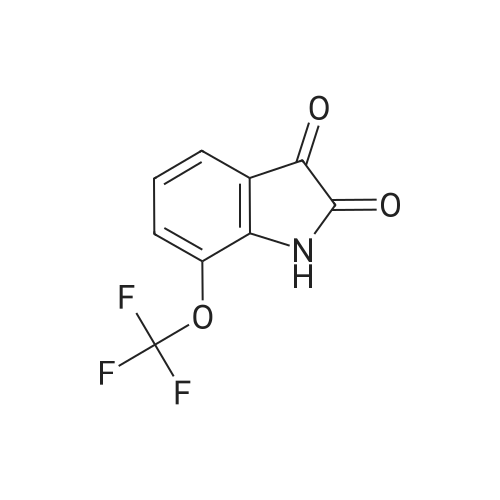
Stage #1: at 65 - 80℃; for 1.5 h; Stage #2: With sodium hydroxide In chloroform; water; isopropyl alcohol at 0℃;
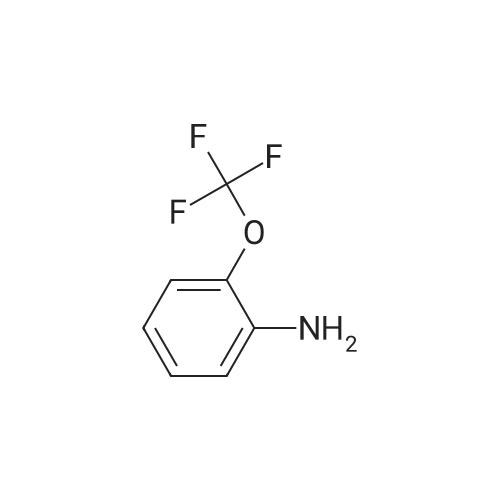
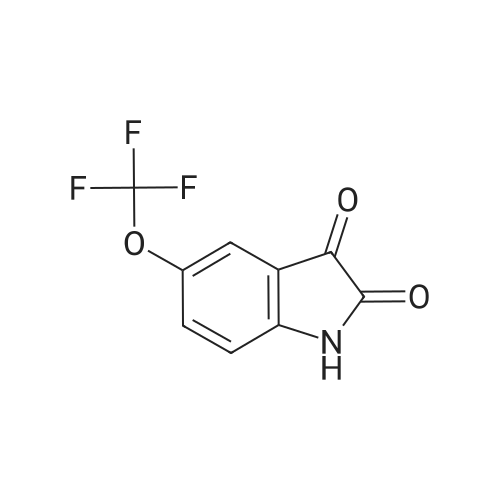
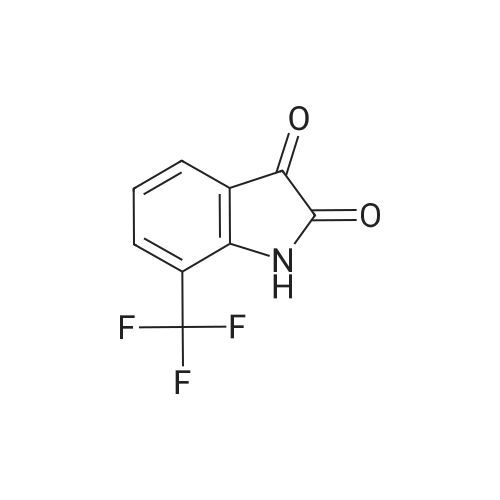
According to Scheme 9, concentrated sulfuric acid (40 mL) was heated to 65° C. and poured into a flask containing the crude 2-(trifluoromethoxy)-α-oximinoanilide from Example 1.1. The mixture was stirred at 65° C. until homogeneous and then the temperature was increased to 80° C. After 90 min, the resulting black mixture was poured into ice/water (300 mL) and diluted with 15percent i-PrOH/CHCl3 (200 mL). After slow addition of 6 N NaOH (80 mL, 15 mins), the aqueous layer was extracted with CHCl3 (2.x.80 mL). To the combined organic layers was added silica gel (30 g) and the solvent was then evaporated. The resulting solid was applied to a column of silica gel and eluted with 15-25percent EtOAc/hexanes to provide the product (5.0 g, 59percent, 2 steps): MS(ESI): 230 (M-H)
Reference:
[1] Chinese Chemical Letters, 2017, vol. 28, # 6, p. 1248 - 1251
[2] Patent: US2008/255192, 2008, A1, . Location in patent: Page/Page column 16-17
[3] Patent: US2005/59823, 2005, A1, . Location in patent: Page 15
2
[ 1535-75-7 ]
[ 149125-30-4 ]
Reference:
[1] Journal of Medicinal Chemistry, 2007, vol. 50, # 1, p. 21 - 39
[2] Journal of Medicinal Chemistry, 2007, vol. 50, # 1, p. 40 - 64
[3] Chinese Chemical Letters, 2017, vol. 28, # 6, p. 1248 - 1251
According to Scheme 10, the starting isatin (3.5 g, 15 mmol) and 6 N NaOH (25 mL, 10 equivalents) were heated at 80 C. and treated slowly with 30% H2O2 (4.5 mL, 0.3 mL/mmol, gas evolution). After 1 h, the mixture was cooled to room temperature and 6 N HCl was added dropwise (27 mL), whereupon a precipitate formed. After 30 mins, the mixture was cooled to 0 C. and the solid was filtered, washed with ice cold water (15 mL) and dissolved in CH2Cl2 (45 mL). The organic solution was dried (Na2SO4), filtered and evaporated to provide the product as an off-white solid (3.4 g, 88%): MS(ESI): 220 (M-H)-.
General procedure: Concentrated sulfuric acid (95 mL) was heated in a flask at 60 C. Intermediates 2 (12 g, 52mmol) were slowly added in portions to maintain the temperature below 70 C. The temperature was then increased to 80 C. After 3 h, the solution was cooled to room temperature, and then poured into crushed ice with vigorous stirring. The precipitate was collected by filtration and dried under vacuum to provide the product 3.
70%
With sulfuric acid; at 55 - 80℃; for 0.166667h;
The procedure described by Marvel et al. (see Org. Synth. Coll. Vol. I, 327) was followed. Intermediate 12 (11.9 g, 48.5 mmol) was added in small portions to 35 mL of concentrated sulfuric acid at 55 C. in a 250 mL Erlenmeyer flask. The temperature of the solution was maintained below 70 C. until all the acetamide had been added and it was increased to 80 C. for 10 minutes. The dark-colored solution was cooled to room temperature and poured onto 175 mL of crushed ice. After standing for 30 minutes, the precipitate was collected by filtration, washed three times with water, and dried under vacuum to yield indoline-2,3-dione of sufficient purity to be used in the next step (intermediate 13, 8.32 g, 70% yield). 1H NMR (400 MHz, DMSO-d6) delta 7.15 (t, J=7.8 Hz, 1H), 7.56 (d, J=7.3 Hz, 1H), 7.64 (d, J=8.3 Hz, 1H), 11.71 (s, 1H).
59%
According to Scheme 9, concentrated sulfuric acid (40 mL) was heated to 65 C. and poured into a flask containing the crude 2-(trifluoromethoxy)-alpha-oximinoanilide from Example 1.1. The mixture was stirred at 65 C. until homogeneous and then the temperature was increased to 80 C. After 90 min, the resulting black mixture was poured into ice/water (300 mL) and diluted with 15% i-PrOH/CHCl3 (200 mL). After slow addition of 6 N NaOH (80 mL, 15 mins), the aqueous layer was extracted with CHCl3 (2×80 mL). To the combined organic layers was added silica gel (30 g) and the solvent was then evaporated. The resulting solid was applied to a column of silica gel and eluted with 15-25% EtOAc/hexanes to provide the product (5.0 g, 59%, 2 steps): MS(ESI): 230 (M-H)
Intermediate 13 (1.00 g, 4.32 mmol) was taken up in 1 mL of ethanol and 3.4 mL of 10 M sodium hydroxide and the resulting mixture was heated at reflux temperature for 20 minutes. A solution of intermediate 55 in 7 mL of ethanol was added dropwise via syringe and the resulting mixture was heated overnight. It was cooled to room temperature and ethanol was removed under reduced pressure. The residue was diluted with water, acidified to pH 1 by slow addition of 1 M hydrochloric acid, and extracted with ethyl acetate. The combined ethyl acetate layers were concentrated to give a dark material which was purified by preparative HPLC (water/acetonitrile with 0.1% triethylamine). The purified triethylammonium salt was taken up in 20% acetonitrile in water and acidified with concentrated hydrochloric acid. Pure product precipitated out of solution as an off-white powder was collected to give Compound 1 (83 mg, 4.5% yield). 1H NMR (400 MHz, DMSO-d6) delta 1.35-1.39 (m, 2H), 1.49-1.54 (m, 2H), 7.16 (d, J=8.6 Hz, 2H), 7.28 (d, J=8.6 Hz, 2H), 7.57 (d, 1H), 7.62 (t, 1H), 8.69 (d, J=7.3 Hz, 1H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping