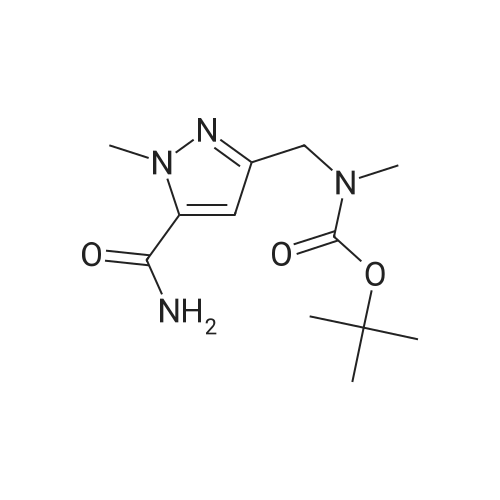
| 95% |
With triethylamine; In ethyl acetate; at 0 - 25℃; for 2h;Product distribution / selectivity; |
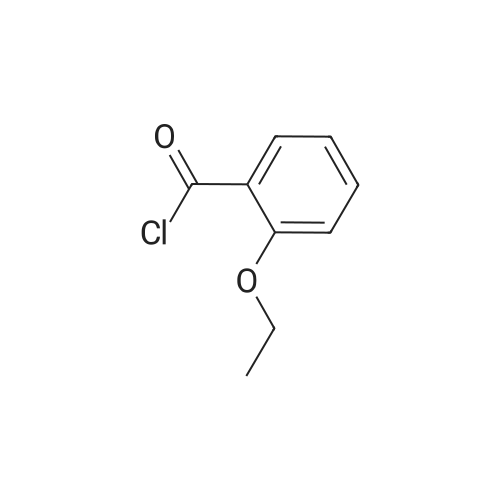
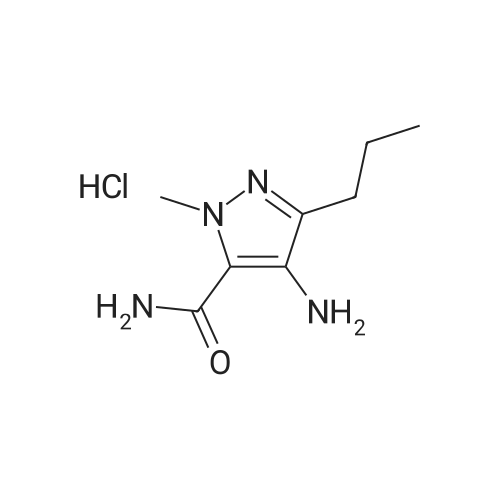
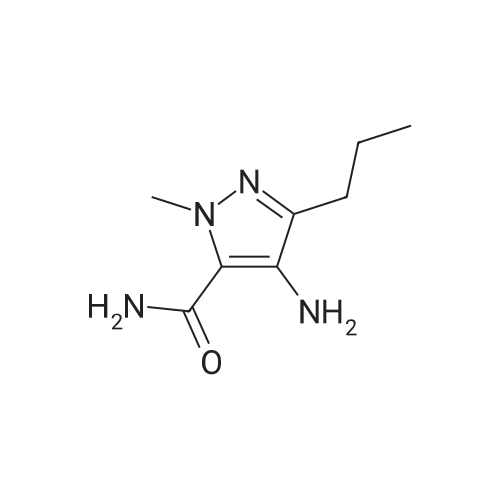
Preparation Ia: l-methyl-3-n-propyl-4- (2-ethoxybenzamido) -pyrazole-5-carbox- amide (V)In a 250 mL three-neck bottle, (XI) (21.8 g, 0.10 mol) was dissolved in ethylacetate (200 mL) and triethylamine (32 ml, 0.23 mol) to prepare a solution in an ice bath. 2- ethoxybenzoyl chloride (17.0 mL, 0.11 mmol) was added to the solution below 5 0C, and the mixture was stirred at room temperature for 2 hours. Water (170 mL) and petroleum ether (100 mL) were added to stop the reaction, and the mixture was stirred for another 0.5 h. The solid was filtrated and poured into water (170 mL) and stirred for 0.5 h, filtrated and dried (70 0C, 12 h) to afford compound (V) as a white solid (32.0 g, yield 95 %) . m.p. 153 - 154 0C. The product was re- crystallized from ethyl acetate/petroleum ether. 1H NMR (CDCI3, 300 MHz) delta: 0.93 (3H, t) , 1.54 (3H, t) , 1.65 (2H, m) , 2.54 (2H, t), 4.06 (3H, s), 4.31 (2H, q) , 5.62 (IH, br s) , 7.05 (IH, d) , 7.13 (IH, t), 7.54 (IH, t) , 7.91 (IH, br s) , 8.27 (IH, dd) , 9.47 (IH, s) . |
| 87% |
With triethylamine; In dichloromethane; at 0 - 25℃; for 2h;Product distribution / selectivity; |
Preparation 1 : l-methyl-3-n-propyl-4- (2-ethoxybenzamido) -pyrazole-5-carbox- amide (V)In a 250 mL three-neck bottle, (XI) (20 g, 0.11 mol) was dissolved in dichloromethane (100 mL) and triethylamine (22.2 g, 0.22 mol) to prepare a solution in an ice bath. 2- ethoxybenzoyl chloride was added to the solution below 5 0C, and the mixture was stirred at room temperature for 2 hours. Water (40 mL) was added to stop the reaction, and the layers were separated. The organic phase was washed with brine (30 mL) and saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, followed by concentration. The resulting residue was purified by re- crystallizing from ethyl acetate/petroleum ether to obtain (V) (31.5 g, yield 87 %) as a white solid, m.p. 153 - 154 0C. 1H NMR (CDCl3, 300 MHz) delta: 0.93 (3H, t), 1.54 (3H, t), 1.65 (2H, m) , 2.54 (2H, t) , 4.06 (3H, s) , 4.31 (2H, q) , 5.62 (IH, br s) , <n="19"/>7 . 05 ( IH, d) , 7 . 13 ( IH, t ) , 7 . 54 ( IH , t ) , 7 . 91 ( IH , br s ) , 8 . 27 ( IH , dd) , 9 . 47 ( IH, s ) . |
| 87% |
With triethylamine; In dichloromethane; at 25℃;Cooling with ice; |
To a 250 mL three-neck flask, were added the compound of formula (XI) (20 g, 0.11 mol), dichloromethane (100 mL) and triethylamine (22.2 g, 0.22 mol) to prepare a solution in an ice bath below 5 C., followed by the slow addition of <strong>[42926-52-3]2-ethoxybenzoyl chloride</strong>. After the mixture was stirred at room temperature for 2 hours, water (40 mL) was added to quench the reaction, and the layers were separated. The organic phase was washed with brine (30 mL) and saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, followed by concentration. The resulting residue was purified by re-crystallising with ethyl acetate/petroleum ether to obtain the compound of formula (V) (31.5 g, yield 87%) as a white solid. m.p. 153154 C.; 1H NMR (CDCl3, 300 MHz) delta: 0.93 (3H, t), 1.54 (3H, t), 1.65 (2H, m), 2.54 (2H, t), 4.06 (3H, s), 4.31 (2H, q), 5.62 (1H, br s), 7.05 (1H, d), 7.13 (1H, t), 7.54 (1H, t), 7.91 (1H, br s), 8.27 (1H, dd), 9.47 (1H, s). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping