* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of the Chemical Society, 1913, vol. 103, p. 2181
[2] Journal of the Chemical Society, 1913, vol. 103, p. 1301
[3] Proceedings of the Chemical Society, London, 1913, vol. 28, p. 333[4] Journal of the Chemical Society, 1913, vol. 103, p. 989
2
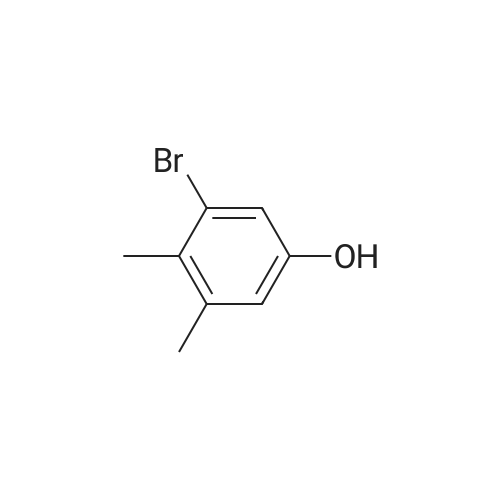
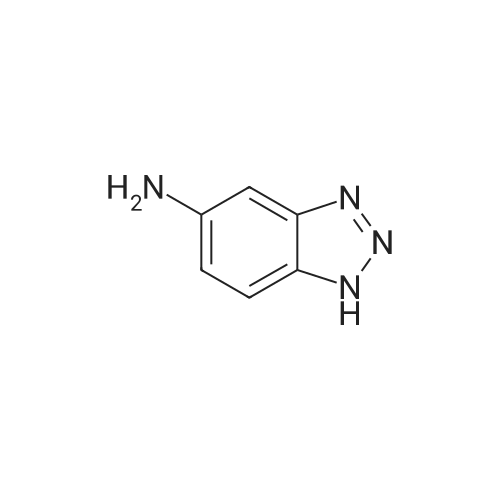
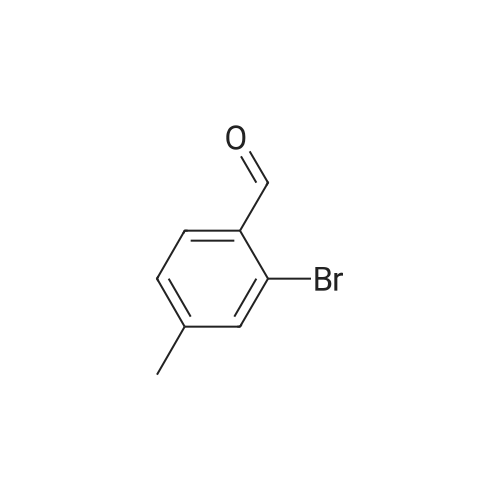
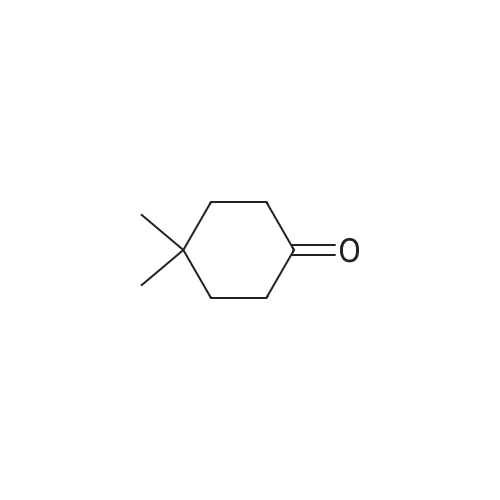
[ 7789-69-7 ]
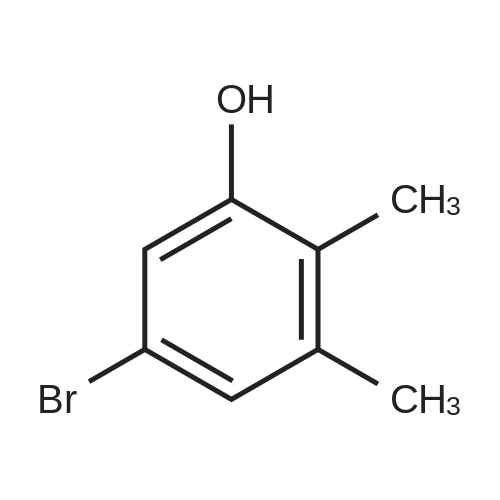
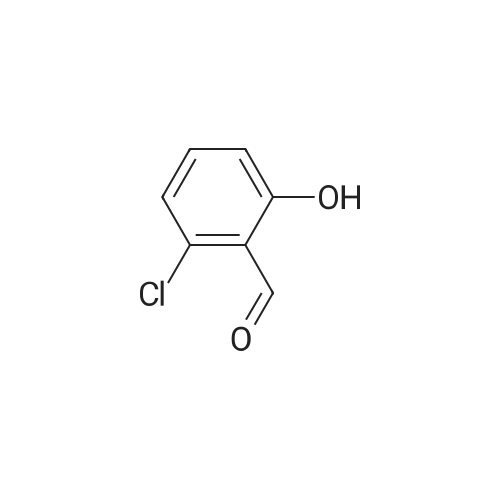
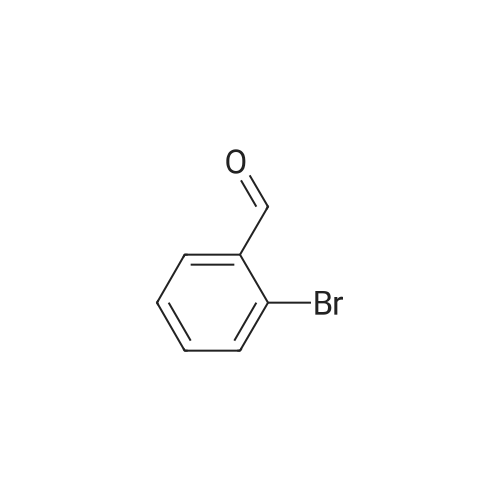
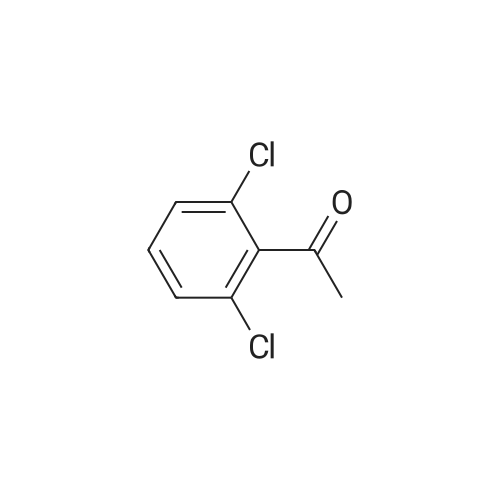
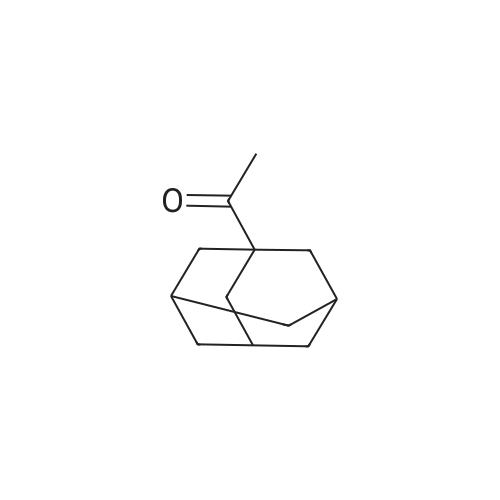
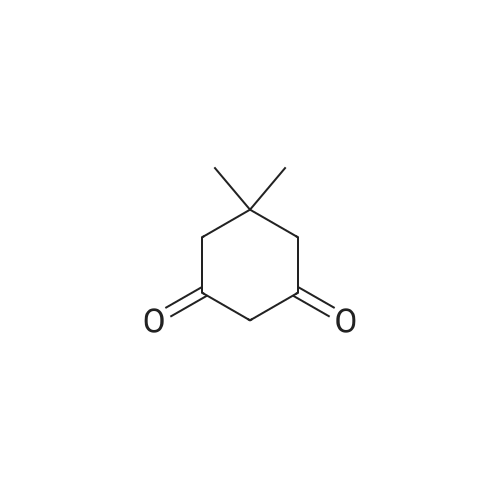
[ 126-81-8 ]
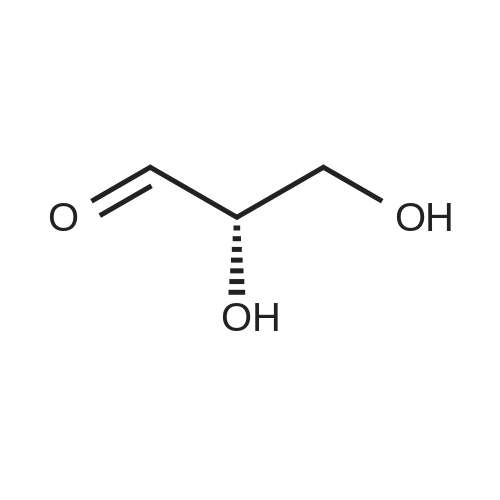
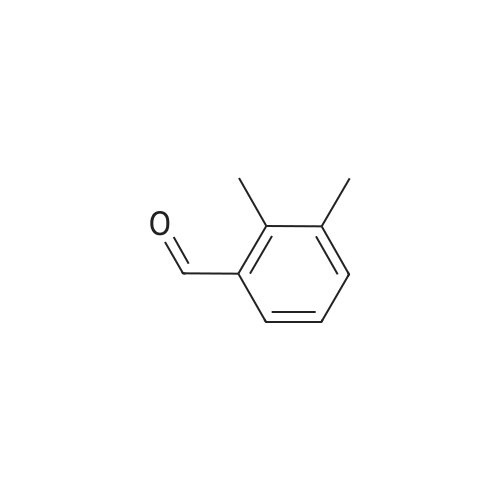
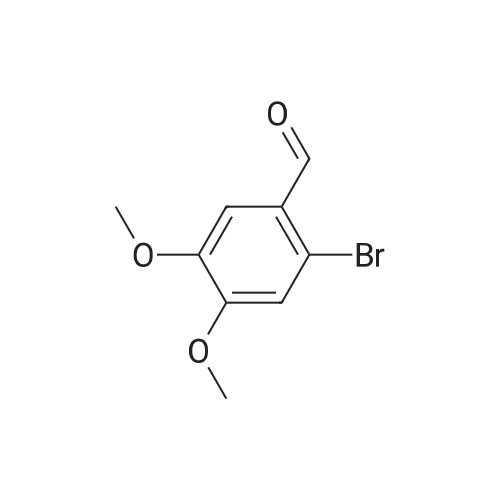
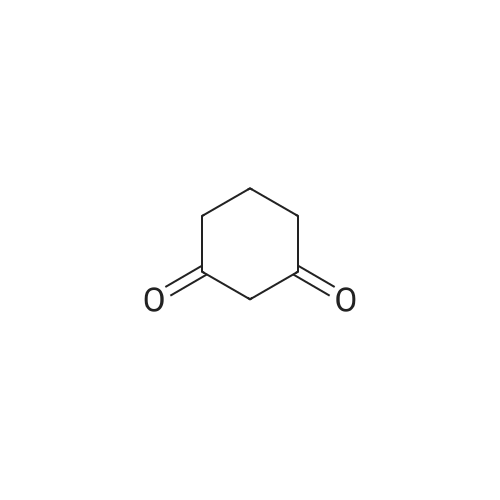
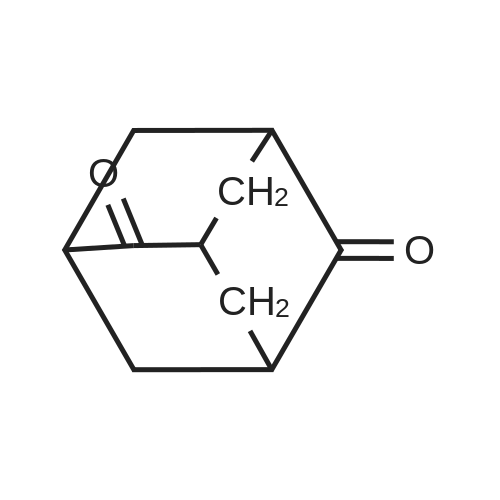
[ 71942-14-8 ]
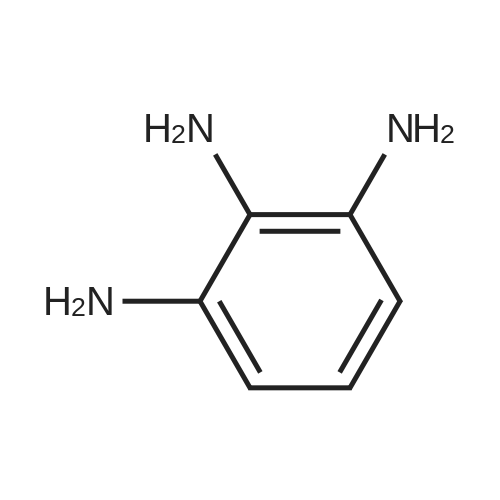
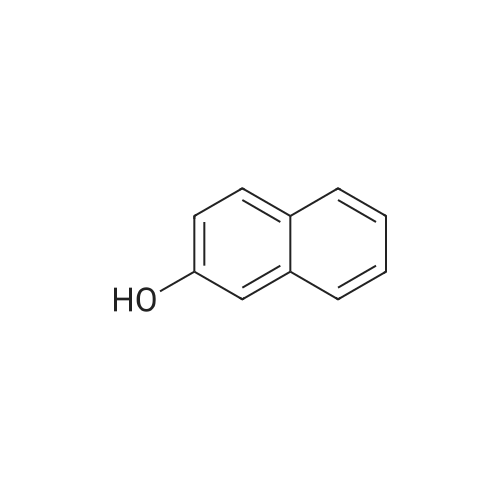
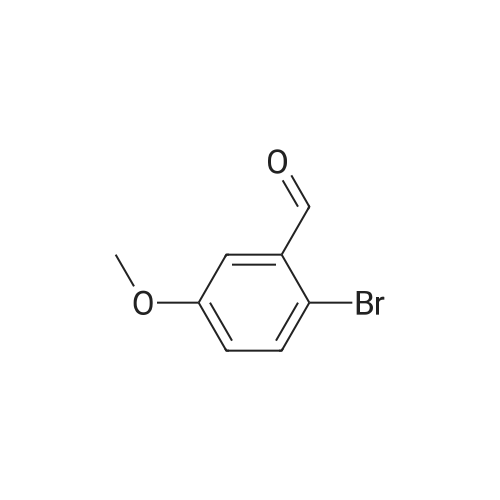
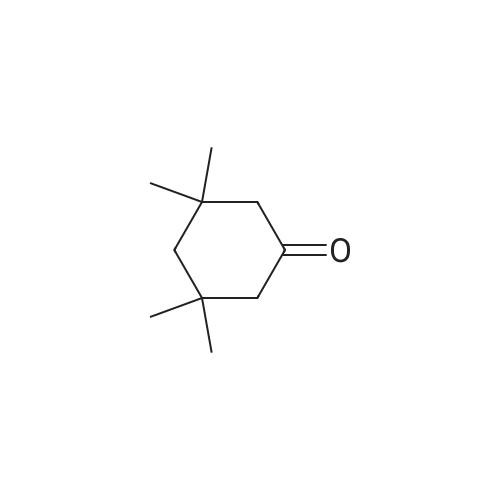
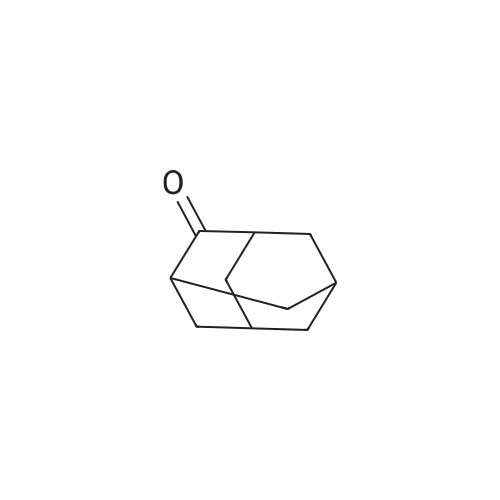
[ 203808-81-5 ]
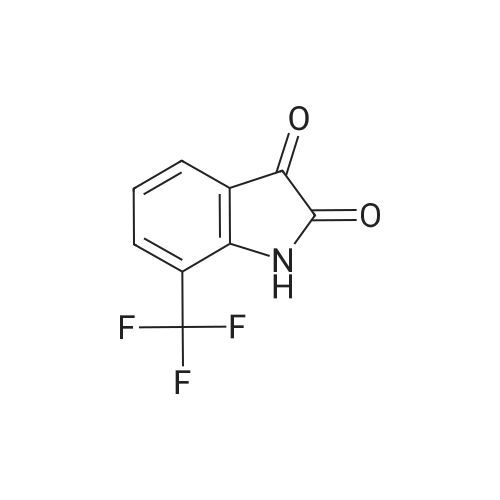
[ 838841-06-8 ]
Reference:
[1] Journal of the Chemical Society, 1913, vol. 103, p. 2181
[2] Journal of the Chemical Society, 1913, vol. 103, p. 1301
[3] Proceedings of the Chemical Society, London, 1913, vol. 28, p. 333[4] Journal of the Chemical Society, 1913, vol. 103, p. 989
In toluene; for 12.0h;Heating / reflux; dean stark apparatus;
A mixture of v-1 (0.0089 mol) and v-2 (0.0089 mol) in toluene (50 ml) was stirred and refluxed for 12 h in a Dean Starck apparatus, then cooled to room temperature. The precipitate was filtered, washed with diethyl ether and dried, yielding: 2 g of v-3 (100percent).A mixture of v-3 (0.0106 mol) and v-4 (0.0106 mol) in AcOH (2.6 ml) and EtOH (50 ml) was stirred at 750C for 24 hours, then cooled to room temperature and concentrated under reduced pressure. The residue was taken up in CH2Cl2. The organic layer was washed with K2C O3 10percent, dried (over MgSO4), filtered and the solvent was evaporated. The residue (5.7 g) was purified by column chromatography over silica gel (eluent: CH2C12/CH3OH/NH4OH 100/0/0 to 99/1/0.1). Three fractions were collected and the EPO <DP n="118"/>solvent was evaporated, yielding: 0.27 g of v-5 (4.5percent) (melting point > 2600C), 0.4 g of v-6 (6.7percent) and 0.34 g of v~7 (5.7percent) (melting point > 2600C).A mixture of v-6 (0.0006 mol) and NH2-NH2/H2O (0.003 mol) in EtOH (20 ml) was stirred and refluxed for 6 hours, then concentrated under reduced pressure. The residue was crystallized from CH3CN. The precipitate was filtered off and dried, yielding: 0.12 g (50percent). Part of this fraction (0.04 g) was crystallized from CH3CN. The precipitate was filtered off and dried, yielding: 0.03 g of v-8 (melting point: 248°C).
With glucose sulfonic acid; In water; at 90℃; for 3h;
General procedure: A mixture of aromatic aldehydes (1, 2.0 mmol), dimedone (2, 2.0 mmol), 2-naphthol or beta-naphthylamine (3 or 4, 2.0 mmol) and 5 molpercent GSA was stirred in water (2 mL) at 90 °C until completely (monitored by TLC). After the completion, water (20?25 mL) was added to quench the reaction and then the reaction mixture was filtered, concentrated, the precipitate was collected, and purified by 95percent EtOH/DMF (8:1).
With glucose sulfonic acid; In water; at 90℃; for 7h;
General procedure: A mixture of aromatic aldehydes (1, 2.0 mmol), dimedone (2, 2.0 mmol), 2-naphthol or beta-naphthylamine (3 or 4, 2.0 mmol) and 5 molpercent GSA was stirred in water (2 mL) at 90 °C until completely (monitored by TLC). After the completion, water (20?25 mL) was added to quench the reaction and then the reaction mixture was filtered, concentrated, the precipitate was collected, and purified by 95percent EtOH/DMF (8:1).
With perchloric acid * acetic acid; In ethanol; water; at 80℃; for 3h;pH 4.4;Green chemistry;
General procedure: A mixture of 5-amimotetrazole (1 mmol, 0.085 g), appropriateisatin (1 mmol), dimedone (1 mmol, 0.14 g) and [MeC(OH)2]+ClO4?(10 molpercent) was stirred in EtOH/H2O (1:1, 4 mL) at 80 °C. After theindicated time, the reaction was quenched into water (50 mL) andcooled. The precipitated product was separated by centrifugation andpurified by silica gel chromatography to afford the desired product. Allthe products were characterised by melting point, 1H NMR, 13C NMRand LC-MS.
General procedure: A mixture of o-phenylenediamine (1mmol), dimedone or1,3-cyclohexanedione (1mmol) and Fe(III)-NicTC(at)nSiO2(3.0molpercent, 30mg) was stirred at room temperature for 20min. Then, aldehyde (1mmol) and water (5mL) wereadded to the reaction mixture and stirred for the stipulated period time. Up to the end of the reaction, the catalyst was separated by centrifuge and the filtrate was concentrated under reduced pressure. After that, the oily mixture was dissolved in ethanol and the pure product was obtained by recrystallization from ethanol (Table2). The synthesis of mono and bis-benzodiazepine derivatives was also performed under the same conditions (Scheme2).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping