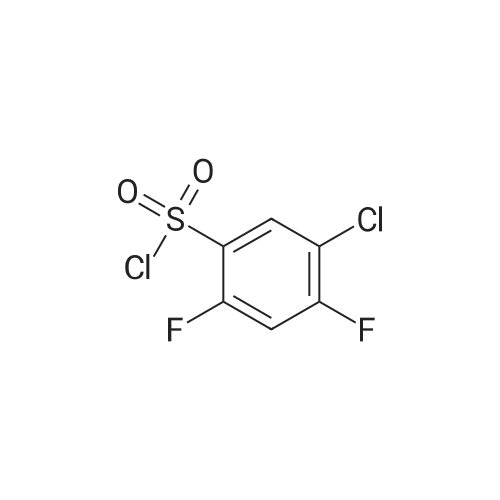
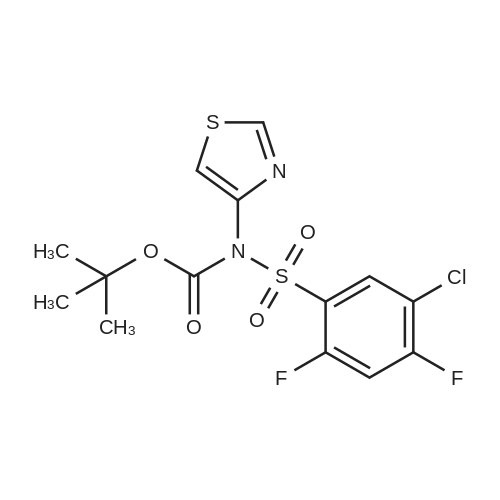
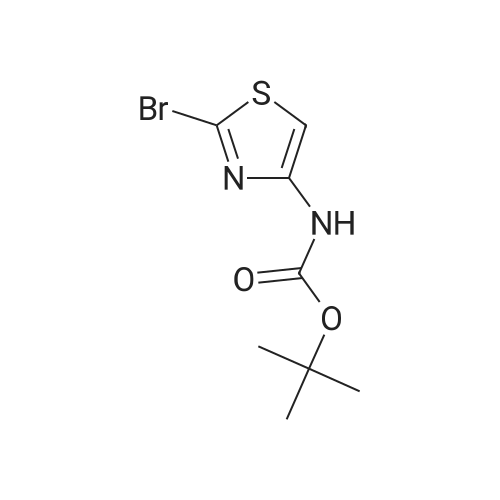
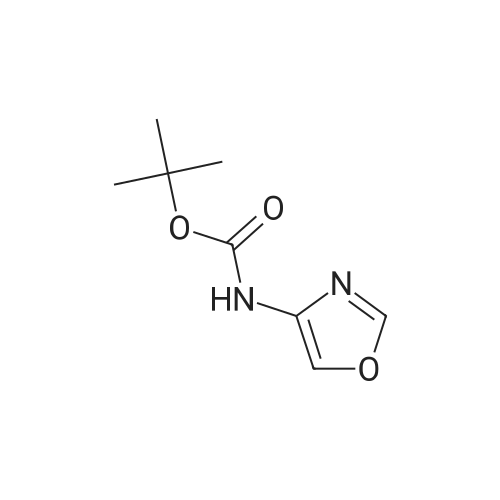
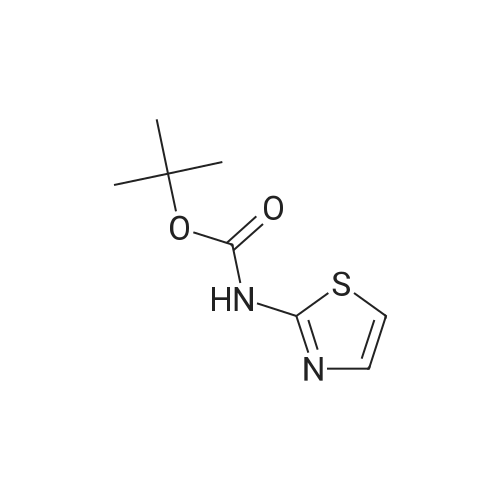
| 60.6% |
With diphenyl phosphoryl azide; triethylamine; at 0 - 90℃; |
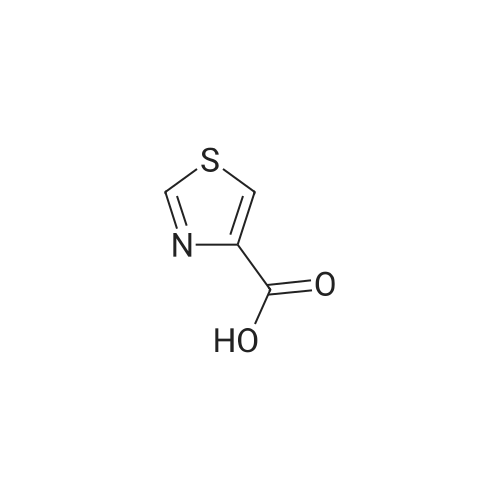
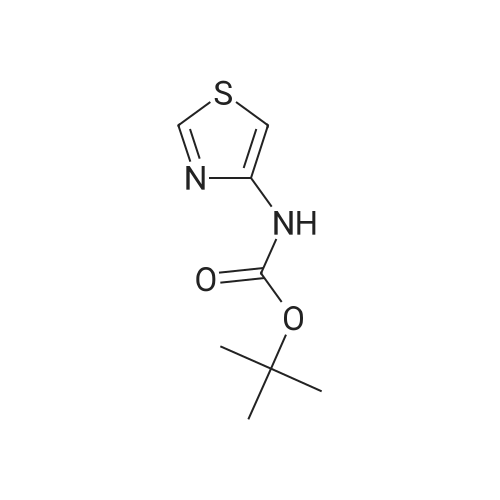
2.1 tert-butyl thiazo 1-4-ylcarbamate DPPA (1.19 g, 4.34 mmol) was added dropwise to a mixture of thiazole-4- carboxylic acid (0.5 g, 3.87 mmol) and triethylamine (0.44 g, 4.30 mmol) in tert-BuOH (50 mL) at 0 C -5 C. The mixture was heated to 90 C for overnight. The solvent was evaporated in vacuo, the residue was diluted with water and extracted with EA (3 x 20 mL). The combined organic phase was washed with brine, dried with Na2S04, concentrated under reduced pressure to give crude product. The crude product was purified by column chromatography on silica gel (EA/heptane=l/10) to give the title compound (0.47 g, 2.35 mmol, 60.6%) as a white solid. LC-MS : m/z 201 (M+H), RT=1.28 min; 1H NMR (400 MHz, CDC13): delta = 8.59 (d, J =2.4 Hz, 2H), 7.31 (s, 1H), 1.54 (s, 9H). |
| 51.5% |
With diphenylphosphoranyl azide; triethylamine; In tert-butyl alcohol; at 90 - 100℃; for 72h; |
Thiazol-4-carboxylic acid (5.0 g, 38.8 mmol) was dissolved in t-BuOH (100 mL), and then TEA (8.1 mL, 1.5 eq.) and DPPA (7.1 mL, 1.5 eq.) were added thereto. While maintaining the internal temperature at 90 to 100C, the reaction mixture was stirred for 3 days, and then the completion of the reaction was confirmed by TLC. The product was concentrated under reduced pressure, distilled water (50 mL) was added, and extracted twice with EA (100 mL). MgSO4 was added to the organic layer, which was stirred, dried and then filtered. The filtrate was concentrated under reduced pressure, and the residue was added to a small amount of EA and slurried. The resulting solid was filtered to obtain a white title compound (4.0 g, 51.5%). 1H NMR (MeOD): 8.73(s, 1H), 7.24(s, 1H), 1.52(s, 9H) |
| 27% |
With diphenyl phosphoryl azide; triethylamine; at 100℃; for 17h; |
A mixture of thiazole-4-carboxylic acid (8.16 g, 63.2 mmol) and triethylamine (9.7 mL, 70 mmol) in tert-butanol (320 mL) was treated with diphenyl phosphoryl azide (15 mL, 70 mmol) and heated gradually in an oil bath to 100 C. and stirred for 17 h. Once cooled, the crude reaction mixture was concentrated under reduced pressure to remove most of the volatiles. The residue was transferred to a 1.0-L separatory funnel with ethyl acetate (300 mL) and the organic layer was washed with water (2×200 mL) and brine (2×100 mL). The organic layer was then dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified silica gel chromatography, eluting from 0% to 50% ethyl acetate in heptanes. The resultant gummy solid was triturated with heptanes, filtered, washed with heptanes and dried under high vacuum to afford tert-butyl thiazol-4-ylcarbamate (3.42 g, 27%) as a white solid. 1H NMR (300 MHz, CDCl3) delta 1.54 (s, 9H), 7.30 (br. s, 1H), 8.33 (br. s, 1H), 8.58 (d, J=2.3 Hz, 1H). [M-C4H8+H]+=145.1. |
|
With diphenyl phosphoryl azide; triethylamine; for 18h;Reflux; |
Thiazole-4-carboxylic acid (6.46 g, 50.0 mmol) was slurried in terf-butyl alcohol (280 mL). Triethylamine (7.68 mL, 55.1 mmol) and diphenylphosphonic azide (1 1 .9 mL, 55.1 mmol) were added and the reaction mixture heated at reflux for 18 hours. The reaction mixture was concentrated in vacuo and the residue dissolved in ethyl acetate. The organics were washed successively with water, 5% aqueous citric acid solution, water, saturated aqueous sodium bicarbonate and brine. The organic phase was dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was purified by silica gel chromatography (ISCO column, hexanes to ethyl acetate gradient elution) then triturated with 20% terf-butyl methyl ether in hexanes. The solid was collected by filtration and dried in vacuo to afford the title compound as a white solid (6.48 g).1 HNMR (CDCIs): delta 1 .46 (s, 9H), 7.23 (m, 1 H), 8.89 (d, 1 H).LCMS Rt = 1 .46 min MS m/z 201 [MH]+ |
|
With diphenylphosphoranyl azide; triethylamine; In tert-butyl alcohol; at 100℃; for 8h;Inert atmosphere; |
[0168] To a solution of 40-1 (8.9 g, 68 mmol), Et3N (7.8 g, 76 mmol) in t-BuOH (100 mL) was added DPPA (21 g, 77 mmol), then the mixture was stirred for 8 h under N2 at 100 C. Aftercooled to room temperature, the solvent was removed in vacuo. The residue was dissolved in CH2CI2 and the organic layer was washed with water and brine, dried over Na2504. The crudeproduct was purified by column chromatography on silica gel (PE: EtOAc = 4:1) to give 40-2. ?H NMR (400 MHz CDCI3) 5 8.87 (brs, 1H), 8.61?8.62 (m, 1H), 7.26?7.27 (m, 1H), 1.54 (s, 9H). |
|
With diphenyl phosphoryl azide; triethylamine; at 100℃; for 8h;Inert atmosphere; |
To a solution of 40-1 (8.9 g, 68 mmol), Et3N (7.8 g, 76 mmol) in t-BuOH (100 mL) was added DPPA (21 g, 77 mmol), then the mixture was stirred for 8 h under N2at 100 C. After cooled to room temperature, the solvent was removed in vacuo. The residue was dissolved in CH2CI2 and the organic layer was washed with water and brine, dried over Na2504. The crude product was purified by column chromatography on silica gel (PE: EtOAc = 4:1) to give 40-2. 1H NMR (400 MHz CDCl3) 5 8.87 (brs, 1H), 8.61?8.62 (m, 1H), 7.26?7.27 (m, 1H), 1.54 (s, 9H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping