| 79% |
With tert.-butylhydroperoxide;bis(acetylacetonate)oxovanadium; In ethanol; water; at 16 - 17℃; for 3.08333h; |
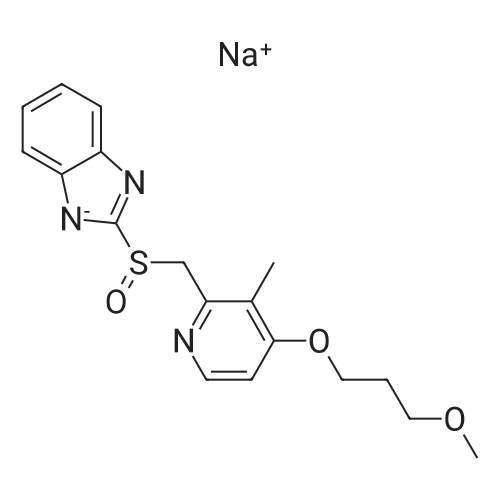
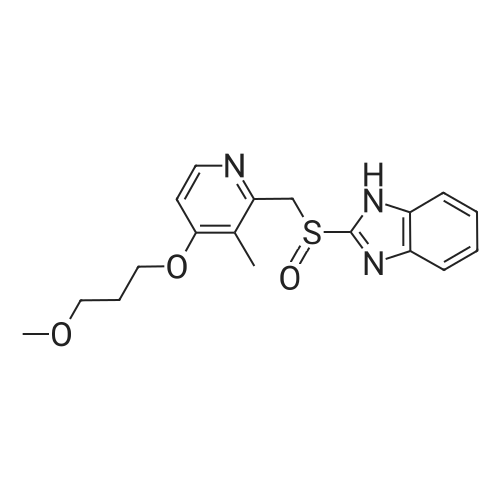
1.5 mg (0.6% molar) VO(acac)2) is dissolved in 12 ml ethanol at room temperature. The solution is stirred and 3 grams of 2-[[[4-(3-methoxy-propxy)-3-methyl-2-pyidinyl]methyl]thio]-1H-benzimidazole are added. 1.5 ml aqueous tert-butyl hydroperoxide (TBHP) (70%) is added over a 5-minute period at 16-17 C. and the solution is then stirred for 3 hours. After completion of the reaction, the product mixture is cooled to about 15 C. and treated with aqueous sodium metabisulphate. The resultant solid is filtered off, washed with cooled ethyl acetate to afford the end product as an almost white solid (2.5 grams, yield 79%). |
| 78.25% |
|
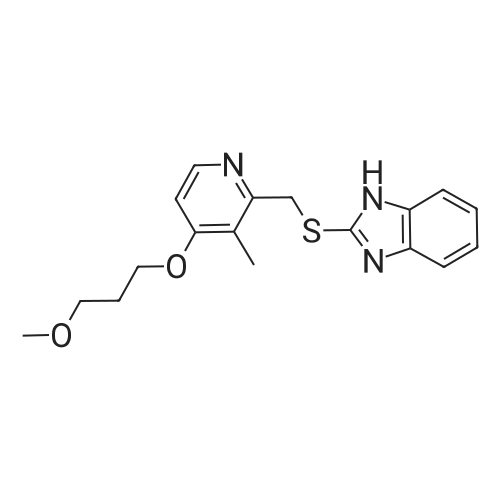
50 g of 2-[4-(3-methoxy propoxy-3-methylpyridine-2-yl) methyl thio]- lH-benzimidazole was taken in 100 ml acetonitrile. To this mixture 185.2 g (0.95mol) sodium hypochlorite was added drop wise between 0-50C in a nitrogen atmosphere and the obtained mixture was stirred at 0-50C for 30 minutes. After completion of reaction, the pH of reaction mixture was adjusted to 9.0 by using 5 % acetic acid. The mixture was extracted with methylene dichloride, and the extract was dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure and dried under vacuum. The obtained residue was dissolved in 20 ml of methylene dichloride followed by t-butyl methyl ether to get the clear solution. Stir the mass for one hour at room temperature to get the precipitate. Filtered the precipitated product and washed with 100 ml of t-butyl methyl ether. Dry the product at 600C under vacuum to obtain title compound 41 g (78.25%) as off white solid |
| 74.94% |
With sodium hypochlorite; sodium hydroxide; In water; acetonitrile; at 0 - 5℃; for 2h; |
Take 50g of <strong>[117977-21-6]rabeprazole sulfide</strong> composition (145.59mmol) was added to a 1L three-necked flask, followed by addition of 500ml of acetonitrile / water mixture (V / V = 2/1), stirred and solid sodium hydroxide was added 11.65g (291.18 mmol).Then cooled to 0-5 , start added dropwise 140.89g 10% NaClO solution (189.27mmol), after the completion of the dropwise addition the reaction was stirred incubated 2h.The reaction gusset fully monitored, 0-5 slowly added dropwise 11.51gNa2S2O3(72.80mmol) in 100ml of water quench unreacted sodium hypochlorite, then with 5% acetic acid adjusted to PH 8-8.5, to precipitate a solid, heat stirred for 2h.Filtered off with suction, the solid in a vacuum oven, 40-45 blast drying 12h.Sample of rabeprazole was weighed 39.22g (109.11mmol), a yield of 74.94%. |
| 70% |
With sodium hydroxide; sodium hypochlorite; tetrabutylammomium bromide; In dichloromethane; water; at 10 - 25℃; for 1.75h;pH 11 - 12; |
Example 1 - Synthesis of 24f4-(3-Methoxypropoxy)3-methylpyridine-2- yllmethylthiol-lH-benzimidazole (Rabeprazole)Example Ia 100 g of Rabeprazole sulphide was dissolved in 500 ml of methylene dichloride at RT and 2.0 g of tetrabutylammonium bromide was added under stirring at RT. The reaction mass was cooled to 10-150C and 418 ml aqueous 5.5 wt% sodium hypochlorite solution was charged slowly for 45 min, and the reaction mixture was stirred for one hour. pH was monitored and kept between 11 and 12 using diluted caustic solution (5% NaOH). The reaction mixture was warmed to 20-250C and stirred for 1-2 hrs.The pH was adjusted to 7.5 to 8.0 with 50% acetic acid. The methylene dichloride layer was separated and the aqueous layer was extracted with 300 ml of methylene dichloride. The solvent was evaporated under vacuum at 35C and 300 ml of acetonitrile was added at room temperature. The resultant mass was heated to 50-550C to dissolve the residue completely, then cooled to 0-50C and maintained at this temperature for 1.0 hour. The compound was filtered and washed with 2 x 25 ml of acetonitrile. Finally, the compound was dried at 40 - 450C under vacuum for 2-3 hrs.The dry product thus obtained weighed 70 - 75 g (70% yield), purity as determined by HPLC was 98.5 - 99 %. |
| 67.2 - 67.4% |
With sodium hydroxide; sodium hypochlorite; In water; acetonitrile; at 0 - 5℃; for 0.166667 - 0.333333h;Product distribution / selectivity; |
EXAMPLE 8: PREPARATION OF RABEPRAZOLE USING 1.0 EQUIVALENT OF SODIUM HYPOCHLORITE AND 2.25 EQUIVALENTS OF SODIUM HYDROXIDE:RAB-1 (20Og) and acetonitrile (600 ml) was taken into a round bottom flask and cooled to 0 to 5 0C. A mixture of a solution of 52.47 g of sodium hydroxide in 240 ml of water and 11.2% aqueous sodium hypochlorite solution (400 ml) and was added to it at the same temperature, and maintained for about 10 minutes. Reaction completion was checked using thin layer chromatography. After the reaction was completed, a solution of sodium thiosulfate (80 g) in water (400 ml) was added to it, and stirred for about 10 minutes. Another water (1000 ml) was added to the reaction mass and the reaction mass was allowed to reach 30 0C. Carbon (20 g) was added to it and stirred for about <n="32"/>15 minutes. Then the reaction mass was filtered and the filtered bed was washed with water (200 ml). The filtrate was washed with toluene (400 ml) in two equal lots and then dichloromethane (400 ml) was added to it. pH of the reaction mass was adjusted to about 8.3 and the organic layer was separated. The aqueous layer was extracted into dichloromethane (60 ml). The combined organic layer was added to methyl tertiary butyl ether (1320 ml) and cooled to about 5 0C. 4 g of seeding material was added to the reaction mass and stirred at about 5 0C for about 3 hours. The separated solid was filtered and washed with methyl tertiary butyl ether (200 ml). The wet material was dried at about 45 to 50 0C for about 4 hours to yield 140.7 g of the title compound (% yield:67.2).Purity by HPLC: 99.7.Impurity 1 (Rabeprazole sulfide): 0.07%.Impurity 2 (Rabeprazole sulfone): 0.04%.EXAMPLE 9: PREPARATION OF RABEPRAZOLE USING 1.0 EQUIVALENT OF SODIUM HYPOCHLORITE AND 2.0 EQUIVALENTS OF SODIUM HYDROXIDE:RAB-1 (25 g) and acetonitrile (75 ml) were taken into a round bottom flask and cooled to 0 to 5 0C. A mixture of a solution of 5.8 g of sodium hydroxide in 30 ml of water and 11.2% aqueous sodium hypochlorite solution (50 ml) and was added to it at the same temperature, and maintained for about 20 minutes. Reaction completion was checked using thin layer chromatography. After the reaction was completed, a solution of sodium thiosulfate (10 g) in water (50 ml) was added to it, and stirred for about 10 minutes. Another lot of water (125 ml) was added to the reaction mass and the reaction mass was allowed to reach 30 0C. Carbon (0.25 g) was added to it and stirred for about 15 minutes. Then the reaction mass was filtered and the filtered bed was washed with water (25 ml). The filtrate was washed with toluene (50 ml) in two equal lots and then dichloromethane (50 ml) was added to it. pH of the reaction mass was adjusted to about 8.4 with acetic acid and the organic layer was separated. The aqueous layer was extracted into dichloromethane (7.5 ml). The combined organic layer was added to methyl tertiary butyl ether (165 ml) and cooled to about 5 0C. 0.5 g of seeding material was added to the reaction mass and stirred at about 5 0C for about 3 hours. The <n="33"/>separated solid was filtered and washed with methyl tertiary butyl ether (25 ml). The wet material was dried at about 45 to 50 0C for about 4 hours to yield 17.6 g of the title compound (% yield: 67.4).Purity By HPLC: 99.6%Impurity 1 (Rabeprazole sulfide): Less than 0.002%.Impurity 2 (Rabeprazole sulfone): 0.02%. |
| 23.5 - 55.4% |
|
Synthesis of 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1H-benzimidazole (Reference Example 7) 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane and cooled. Then, mcpba (70.2% purity; 5.37 g (21.8 mmol)) was added gradually so that the internal temperature did not exceed -15C. 10% Aqueous sodium hydroxide solution (70.8 ml) was then added, and after stirring and still standing the water layer was separated. The separated water layer was washed with dichloromethane (48 ml) twice. After addition of a 2N-ammoriium acetate aqueous solution, the water layer was extracted with dichloromethane (48 ml) twice. The dichloromethane layer was washed with water (48 ml) twice, concentrated under a reduced pressure, crystallized with dichloromethane (14 ml) and acetonitrile (92 ml), and filtered to obtain 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1H-benzimidazole (6.26 g) (HPLC purity 99.7%, yield 23.9%). (Reference Example 8) 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane and cooled. Then, mcpba (70.2% purity; 5.37 g (21.8 mmol)) was added gradually so that the internal temperature did not exceed -15C. 10% Aqueous sodium hydroxide solution (70.8 ml) was then added, and after stirring and still standing the water layer was separated. The separated water layer was washed with dichloromethane (48 ml) twice. After addition of a 2N-ammonium acetate aqueous solution, the water layer was extracted with dichloromethane (48 ml) twice. The dichloromethane layer was washed with water (48 ml) twice, concentrated under a reduced pressure, crystallized with ethyl acetate (66 ml) and filtered to obtain 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1H-benzimidazole (6.15 g) (HPLC purity 99.8%, yield 23.5%).(Reference Example 9) 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane and cooled. Then, mcpba (70.2% purity; 7.16 g (29.1 mmol)) was added gradually so that the internal temperature did not exceed -15C. 10% Aqueous sodium hydroxide solution (70.8 ml) was then added, and after stirring and still standing the water layer was separated. The separated water layer was washed with dichloromethane (48 ml) twice. After addition of a 2N-ammonium acetate aqueous solution, the water layer was extracted with dichloromethane (48 ml) twice. The dichloromethane layer was washed twice with water (48 ml), concentrated under a reduced pressure, crystallized with dichloromethane (18 ml) and acetone (120 ml), and filtered to obtain 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1H-benzimidazole (8.56 g) (HPLC purity 99.7%, yield 32.7%).(Reference Example 10) 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane and cooled. Then, mcpba (70.2% purity; 7.16 g (29.1 mmol)) was added gradually so that the internal temperature did not exceed -15C. 10% Aqueous sodium hydroxide solution (70.8 ml) was then added, and after stirring and still standing the water layer was separated. The separated water layer was washed with dichloromethane (48 ml) twice. After addition of a 2N-ammonium acetate aqueous solution, the water layer was extracted with dichloromethane (48 ml) twice. The dichloromethane layer was washed with water (48 ml) twice, concentrated under a reduced pressure, crystallized with isopropyl alcohol (88 ml), and filtered to obtain 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1H-benzimidazole (8.29 g) (HPLC purity 99.7%, yield 31.7%).(Reference Example 11) 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1 H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane and cooled. Then, mcpba (70.2% purity; 7.16 g (29.1 mmol)) was added gradually so that the internal temperature did not exceed -15C. 10% Aqueous sodium hydroxide solution (70.8 ml) was then added, and after stirring and still standing the water layer was separated. The separated water layer was washed with dichloromethane (48 ml) twice. After addition of a 2N-ammonium acetate aqueous solution, the water layer was extracted with dichloromethane (48 ml) twice. The dichloromethane layer was washed with water (48 ml) twice, concentrated under a reduced pressure, crystallized with acetonitrile (132 ml), and filtered to obtain 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1H-benzimidazole (8.25 g) (HPLC purity 99.7%, yield 31.5%).(Reference Example 12) 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane and cooled. Then, mcpba (70.2% purity; 8.95 g (36.4 mmol)) was added gradually so that the internal temperature did not exceed -15C. 10% Aqueous sodium hydroxide solution (70.8 ml) was then add... |
| 23.5 - 55.4% |
|
Reference Example 7; 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane, and then the solution was cooled, and mcpba (70.2% purity; 5.37 g (21.8 mmol)) was added little by little such that the internal temperature did not exceed -15 C. After the addition, a 10% sodium hydroxide aqueous solution (70.8 ml) was added, the mixture was stirred and then left to stand, and then the aqueous layer was separated off. The separated aqueous layer was washed twice with dichloromethane (48 ml). A 2N ammonium acetate aqueous solution was then put into the solution, and then extraction was carried out twice with dichloromethane (48 ml). The dichloromethane layers were washed twice with water (48 ml), vacuum concentration was carried out, crystallization was carried out using dichloromethane (14 ml) and acetonitrile (92 ml), and then filtration was carried out, thus obtaining 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1H-benzimidazole (6.26 g) (HPLC purity 99.7%, yield 23.9%).; Reference Example 8; 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane, and then the solution was cooled, and mcpba (70.2% purity; 5.37 g (21.8 mmol)) was added little by little such that the internal temperature did not exceed -15 C. After the addition, a 10% sodium hydroxide aqueous solution (70.8 ml) was added, the mixture was stirred and then left to stand, and then the aqueous layer was separated off. The separated aqueous layer was washed twice with dichloromethane (48 ml). A 2N ammonium acetate aqueous solution was then put into the solution, and then extraction was carried out twice with dichloromethane (48 ml). The dichloromethane layers were washed twice with water (48 ml), vacuum concentration was carried out, crystallization was carried out using ethyl acetate (66 ml), and then filtration was carried out, thus obtaining 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1H-benzimidazole (6.15 g) (HPLC purity 99.8%, yield 23.5%).; Reference Example 9; 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane, and then the solution was cooled, and mcpba (70.2% purity; 7.16 g (29.1 mmol)) was added little by little such that the internal temperature did not exceed -15 C. After the addition, a 10% sodium hydroxide aqueous solution (70.8 ml) was added, the mixture was stirred and then left to stand, and then the aqueous layer was separated off. The separated aqueous layer was washed twice with dichloromethane (48 ml). A 2N ammonium acetate aqueous solution was then put into the solution, and then extraction was carried out twice with dichloromethane (48 ml). The dichloromethane layers were washed twice with water (48 ml), vacuum concentration was carried out, crystallization was carried out using dichloromethane (18 ml) and acetone (120 ml), and then filtration was carried out, thus obtaining 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1H-benzimidazole (8.56 g) (HPLC purity 99.7%, yield 32.7%).; Reference Example 10; 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane, and then the solution was cooled, and mcpba (70.2% purity; 7.16 g (29.1 mmol)) was added little by little such that the internal temperature did not exceed -15 C. After the addition, a 10% sodium hydroxide aqueous solution (70.8 ml) was added, the mixture was stirred and then left to stand, and then the aqueous layer was separated off. The separated aqueous layer was washed twice with dichloromethane (48 ml). A 2N ammonium acetate aqueous solution was then put into the solution, and then extraction was carried out twice with dichloromethane (48 ml). The dichloromethane layers were washed twice with water (48 m), vacuum concentration was carried out, crystallization was carried out using isopropyl alcohol (88 ml), and then filtration was carried out, thus obtaining 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1H-benzimidazole (8.29 g) (HPLC purity 99.7%, yield 31.7%).; Reference Example 11; 2-[{4-(3-Methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole (25.0 g (72.8 mmol)) was dissolved in dichloromethane, and then the solution was cooled, and mcpba (70.2% purity; 7.16 g (29.1 mmol)) was added little by little such that the internal temperature did not exceed -15 C. After the addition, a 10% sodium hydroxide aqueous solution (70.8 ml) was added, the mixture was stirred and then left to stand, and then the aqueous layer was separated off. The separated aqueous layer was washed twice with dichloromethane (48 ml). A 2N ammonium acetate aqueous solution was then put into the solution, and then extraction was carried out twice with dichloromethane (48 ml). The dichloromethane layers were washed twice with wate... |
|
With 3-chloro-benzenecarboperoxoic acid; In chloroform; dimethyl sulfoxide; at -15 - -10℃; for 1.5 - 2.5h; |
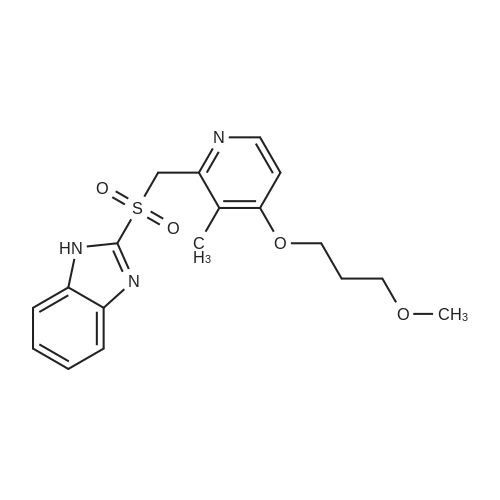
2-[[4-(3-methoxypropoxy)-3-methyl-2-pyridinyl]-methyl thio]-1H-benzimidazole (Prepared as per example 90 of the Patent No. 5,045,552) (100 grams, 0.29 moles) was added to a mixture of chloroform (9500 ml) and dimethylsulphoxide (200 ml) and the reaction mixture was cooled to -10C to -15C. 3-Chloroperbenzoic acid (60 grams, 0.24 moles) was dissolved in chloroform (500ml), and the chloroform solution was then added to the above solution at -10C to -15C over about 1- 2 hours, and the reaction mixture was maintained at the same temperature for 30 minutes. Then, 12.8 % (w/v) aqueous sodium hydroxide solution (500 ml) was added to the reaction mixture. The pH of the reaction mixture was adjusted to 9.5 to 10.0 with acetic acid, which formed a biphasic system. The organic layer was separated and then extracted with 1.6 % (w/v) aqueous sodium hydroxide solution (500 ml). The sodium hydroxide extract was diluted with a mixture of chloroform (140 ml) and methanol (100 ml). Then the pH of the mass was again adjusted to 9.5 to 10.0 with acetic acid, and the organic layer was separated again. To the separated organic layer was added tert. butyl methyl ether (440 ml). The reaction mixture was stirred for about 1-2 hours at a temperature of 0-5 C and was subjected to filtration. The residue was dissolved in a mixture of 110 % (w/v) aqueous sodium hydroxide solution (100 ml) and methanol (65 ml). The pH of the reaction solution was adjusted to 9.0 to 9.5 with acetic acid at 10-15 C and was stirred for additional 2 hours, followed by filtration. The wet material is then dissolved in dichloromethane (130 ml), and the water layer separated where after solution was added to tert. butyl methyl ether (260 ml), stirred at a temperature of 0-5C for 1-2 hours. The precipitated solid was filtered and dried to give 2-[[[4-(3-methoxypropoxy)-3-methyl-2-pyridinyl]-methyl] sulfinyl]-1H-benzimidazole. |
|
With oxone; sodium hydrogencarbonate;bis(acetylacetonate)oxovanadium; In methanol; water; at -2 - 0℃; for 5.5h; |
A mixture of 3 grams 2-[[[4-(3-methoxy-propoxy)-3-methyl-2-pyridinyl]methyl]thio]-1H-benzimidazole, 3 grams NaHCO3 and 20 ml aqueous methanol is cooled to -2 C. and 3.5 ml (5.69 mmol) Oxone is added. The mixture is stirred for 4 hours at 0 C. and a further 1 gram (mmol) Oxone is added and stirring continued for 1.5 hours. A solution of 0.8 gram sodium metabisulfite in 20 ml water is added dropwise over 5-10 minutes. After further stirring the resultant precipitate is filtered, washed successively with water and 50% aqueous methanol and dried. Purity is 98.1%. |
|
With 3-chloro-benzenecarboperoxoic acid; In chloroform; dimethyl sulfoxide; at -15 - -10℃; for 1.5 - 2.5h; |
REFERENCE EXAMPLE 1 Preparation of 2-[[[4-(3-methoxypropoxy)-3-methyl-2-pyridinyl]-methyl]sulfinyl]-1H-benzimidazole (Rabeprazole) 2-[[4-(3-methoxypropoxy)-3-methyl-2-pyridinyl]-methyl thio]-1H-benzimidazole (Prepared as per example 90 of the U.S. Pat. No. 5,045,552) (100 grams, 0.29 moles) was added to a mixture of chloroform (9500 ml) and dimethylsulphoxide (200 ml) and the reaction mixture was cooled to -10 C. to -15 C. 3-Chloroperbenzoic acid (60 grams, 0.24 moles) was dissolved in chloroform (500 ml), and the chloroform solution was then added to the above solution at -10 C. to -15 C. over about 1-2 hours, and the reaction mixture was maintained at the same temperature for 30 minutes.Then, 12.8% (w/v) aqueous sodium hydroxide solution (500 ml) was added to the reaction mixture.The PH of the reaction mixture was adjusted to 9.5 to 10.0 with acetic acid, which formed a biphasic system.The organic layer was separated and then extracted with 1.6% (w/v) aqueous sodium hydroxide solution (500 ml).The sodium hydroxide extract was diluted with a mixture of chloroform (140 ml) and methanol (100 ml).Then the PH of the mass was again adjusted to 9.5 to 10.0 with acetic acid, and the organic layer was separated again.To the separated organic layer was added tert.butyl methyl ether (440 ml).The reaction mixture was stirred for about 1-2 hours at a temperature of 0-5 C. and was subjected to filtration.The residue was dissolved in a mixture of 110% (w/v) aqueous sodium hydroxide solution (100 ml) and methanol (65 ml).The PH of the reaction solution was adjusted to 9.0 to 9.5 with acetic acid at 10-15 C. and was stirred for additional 2 hours, followed by filtration.The wet material is then dissolved in dichloromethane (130 ml), and the water layer separated where after solution was added to tert.butyl methyl ether (260 ml), stirred at a temperature of 0-5 C. for 1-2 hours.The precipitated solid was filtered and dried to give 2-[[[4-(3-methoxypropoxy)-3-methyl-2-pyridinyl]-methyl] sulfinyl]-1H-benzimidazole. |
|
With ammonium acetate; sodium hydroxide; N-chloro-succinimide; sodium chloride; sodium thiosulfate; In hexane; water; ethyl acetate; N,N-dimethyl-formamide; toluene; |
Example 13 Synthesis of 2-[4-(3-Methoxypropoxy)-3-methylpyridin-2-yl]methylsulfinyl}-1H-benzimidazole (Rabeprazole free base) 2-[4-(3-Methoxypropoxy)-3-methylpyridin-2-yl]methylthio}-1H-benzimidazole (5.0 g 14.6 mmol; referred to as Compound I hereinafter) was dissolved in 20 ml of N,N-dimethylformamide, followed by the addition of a 2N aqueous solution of sodium hydroxide (18 ml). A solution (10 ml) of N-chlorosuccinimide (2.71 g, 20.3 mmol) in N,N-dimethylformamide was added dropwise to the solution at -20 C. to -10 C. The reaction mixture was reacted at -15 C. to -7 C. for 1.5 hr. To the reaction mixture was added a 10% aqueous solution of sodium thiosulfate (5 ml), stirred for 2 min and then, a solution (60 ml) of ammonium acetate (23.1 g) in water was added thereto. 60 ml of ethyl acetate and 10 g of sodium chloride were added thereto to extract the reaction mixture, and the aqueous layer was further extracted with ethyl acetate (40 ml). The organic layers were combined, washed with a 15% aqueous solution of sodium chloride (80 ml) for three times and then, the solvent was evaporated. To the resulting oil was added 12 ml of ethyl acetate, 28 ml of hexane and 10 ml of toluene, followed by stirring at room temperature for 1 hr. The resulting crystals were collected by filtration, washed with a solvent mixture (10 ml) of 30% ethyl acetate and hexane for two times and then, dried under reduced pressure, to give the title compound (4.7 g, yield; 90.6%) as grayish white crystals. 1H-NMR (400 MHz, DMSO-d6); delta (ppm) 1.10(t,J=7.2 Hz, 3H), 2.13(s, 3H), 3.50(q,J=7.2 Hz, 2H), 3.71(m, 2H), 4.16(m, 2H), 4.70(d,J=13.6 Hz, 1H), 4.78(d,J=13.6 Hz, 1H), 6.96(d,J5.6 Hz, 1H), 7.28(m, 2H), 7.62(m, 2H), 8.20(d,J=5.6 Hz, 1H). |
|
With ammonium acetate; sodium hydroxide; sodium hydrogencarbonate; triethylamine; 3-chloro-benzenecarboperoxoic acid; In diethyl ether; dichloromethane; |
EXAMPLE 32 2-{4-(3-Methoxypropoxy)-3-methylpyridine-2-yl}methylsulfinyl-1H-benzimidazole STR94 5 g of 2-[{4-(3-methoxypropoxy)-3-methylpyridine-2-yl}methylthio]-1H benzimidazole was dissolved in a mixture comprising 100 ml of dichloromethane and 25 ml of diethyl ether to obtain a solution. 2.83 g of 85% m-chloroperbenzoic acid was added to this solution in portions at -45 C. After the completion of the reaction, 2 g of triethylamine was added to the reaction mixture and the obtained mixture was heated to -10 C., followed by the addition of 50 ml of 1N sodium hydroxide. The obtained mixture was stirred at a room temperature for 30 minutes. The obtained aqueous layer was washed with 20 ml of dichloromethane twice and adjusted to pH 11 with a 2M aqueous solution of ammonium acetate. The aqueous layer was extracted with 50 ml of dichloromethane thrice. The obtained dichloromethane layer was washed with 50 ml of a saturated aqueous solution of sodium hydrogencarbonate twide, dried over magnesium sulfate and distilled to remove the dichloromethane. The obtained oily product was crystallized from dichloromethane/ether to obtain 4.17 g of the title compound as a white crystal. M.p.: 99 to 100 C. (dec.). 1 H-NMR(CDCl3) delta; 1.83~2.09(m, 2H), 2.13(s, 3H), 3.34(s, 3H), 3.52(t, J=6.2 Hz, 2H), 4.05(t, J=6.2 Hz, 2H), 4.79(s, 2H), 6.70(d, J=5.7 Hz, 1H), 7.07~7.30(m, 2H), 7.30~7.60(br, s, 2H), 8.27(d, J=5.7 Hz, 1H). |
|
|
To a mixed solution of 2-hydroxymethyl-4-(3-methoxypropoxy)-3-methylpyridine (12.02 g (56.9 mmol)) and toluene (96.0 ml) was added dropwise thionyl chloride (8.11 g (68.2 mmol)), so that the internal temperature did not exceed 25C, which was stirred for about 90 minutes at room temperature. Ethanol (24.0 ml) was added to this mixed solution to obtain a 2-chloromethyl-4-(3-methoxypropoxy)-3-methylpyridine solution. To this 2-chloromethyl-4-(3-methoxypropoxy)-3-methylpyridin solution was added 2-benzimidazole thiol (8.71 g (58.0 mmol)) at room temperature. A 25% aqueous sodium hydroxide solution (40.6 g) was added gradually as the temperature (internal temperature) was gradually raised to 65C. The reaction mixture was stirred for about an hour and a half at 65C (internal temperature). Water (60.0 ml) was added to the reaction mixture at 65C, after which 25% aqueous sodium hydroxide solution (0.2 g) was added and stirred. This reaction mixture was allowed to stand, and the water layer was separated. The organic layer was washed with water (20.0 ml) twice, and toluene (79.6 ml) and methanol (21.5 ml) were added to the organic layer to obtain a 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole solution. This 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylthio]-1H-benzimidazole solution was cooled to 30C (external temperature), and a solution of mcpba (70.2% purity; 14.39 g (58.5 mmol)), methanol (12.4 ml) and toluene (10.5 ml) were added over the course of about 1 hour so as not to exceed an internal temperature of -25C, and then stirred for an hour and a half. 25% Aqueous sodium hydroxide solution (22.73 g) and water (17.6 ml) were added to the reaction mixture and stirred. This reaction mixture was allowed to stand, and the organic layer was separated to obtain an aqueous alkali solution containing 2-[{4-(3-methoxypropoxy)-3-methylpyridin-2-yl}methylsulfinyl]-1 H-benzimidazole. |
|
|
A suspension of 2-[[[4-(3-Methoxypropoxy)-3-methyl-2-pyridinyl]methyl]tliio]- lH-benzimidazoIe (100 gm, 0.291 moles) Sodium carbonate (30.89 gm, 0.29 moles) and Dimethyl sulphoxide (22.73 gm, 0.291 moles) in chloroform (500 mL) was treated with a solution of peracetic acid (33.2 gm, 0.43 moles) in chloroform at -100C. The reaction mixture was stirred at same temperature for 30 minutes. After completion of reaction, lhe reaction was quenched with sodium thiosulphite and then pH was adjusted to 8.5 - 9.0 with aqueous sodium hydroxide solution and stirred for 15 minutes at 0-50C and organic layer separated. Then the organic layer was back extracted with sodium hydroxide solution at pH 13-14. The pH of aqueous layer thus obtained re-adjusted to 8.0- 8.5 by using acetic acid at 5-1O0C. The precipitate obtained was filtered to give title compound (50 gm). |
|
|
Reference Example; 2- [ {4-(3-methoxypropoxy)-3-methylpyridine-2-yl} methylthio]-1 H- benzimidazole (prepared as per example 90 of the U. S. Patent No. 5,045, 552) (100 grams, 0.29 moles) is added to a mixture of chloroform (500 ml) and dimethylsulfoxide (200 ml) and the reaction mixture is cooled to-10 to-15C. 3-chloroperbenzoic acid (60 grams, 0.24 moles) is dissolved in chloroform (500 ml), and added to the above solution at-10 to - 15C for about 1-12 hours and the reaction mixtures maintained at the same temperature for 30 minutes. Thereafter 12.8% w/v aqueous sodium hydroxide solution (500 ml) is added to the reaction mixture. The pH of the reaction mixture is adjusted to 9.5 to 10.0 with acetic acid. Of the biphasic system thus obtained the organic layer is separated and then extracted with 1.6% w/v aqueous sodium hydroxide solution (500 ml). Further the sodium hydroxide extract is diluted with a mixture of chloroform (140 ml) and methanol (100 ml). Then the pH of the mass is again adjusted to 9.5 to 10.0 with acetic acid and the organic layer separated again. To the separated organic layer is now added tert. butyl methyl ether (440 ml). The reaction mixture is stirred for about 1-12 hours at a temperature of 0-5C and subjected to filtration. The residue is dissolved in a mixture of 10% w/v aqueous sodium hydroxide solution (100 ml) and methanol (65 ml). The pH is adjusted to 9.0 to 9.5 with acetic acid at 10-15C and further stirred for 12 hours followed by filtration. The wet material is then dissolved in dichloromethane (130 ml) and the water layer separated where after the solution is added to tert. butyl methyl ether (260 ml), stirred at a temperature of 0-5C for 1-2 hours. The 2- [ [ [4- (3-methoxypropoxy)-3- methyl-2-pyridinyl]-methyl] sulfinyl]-lH-benzimidazole thus obtained is filtered and dried. |
|
With 3-chloro-benzenecarboperoxoic acid; In chloroform; dimethyl sulfoxide; at -15 - -10℃; for 1.83333 - 2.25h;Product distribution / selectivity; |
2-[[[4-(3-methoxypropoxy)-3-methyl-2-pyridinyl] methyl] thio]-H-benzimidazole compound of Formula III (100 g) was charged into a clean and dry 4 neck round bottom flask containing dimethylsulfoxide (200 ml), and chloroform (500 ml) and stirred for about 10 min. The reaction mixture was cooled to a temperature of about -10 0C to about -15 0C. Metaperoxychlorobenzoic acid (60 g) dissolved in <n="21"/>chloroform (500 ml) was added slowly over about 90 to about 105 minutes at a temperature of about -10 0C to about -15 0C. The resultant reaction mixture was stirred for about 20 minutes to about 30 minutes followed by decomposition of reaction mass by the addition of basic water (500 ml) (64 g sodium hydroxide dissolved in 500 ml of water). The pH of the reaction solution was adjusted to about 8.5 by addition of acetic acid (63 ml) followed by separation of organic and aqueous layers. The aqueous layer was extracted with chloroform (100 ml) followed by separation of the organic and aqueous layers.The combined organic layers were extracted with basic water (300, 200 ml) (5 g of sodium hydroxide dissolved in 300 ml water, 3 g of sodium hydroxide dissolved in 200 ml) followed by washing the aqueous layer with chloroform (2x50 ml). The combined aqueous layer was charged into a clean and dry round bottom flask. Activated charcoal carbon (2g) was added into the reaction mass followed by stirring for about 25 minutes to about 30 minutes. The resultant reaction suspension was filtered through celite and the celite was washed with of water (100 ml).To the resultant clear filtrate, methanol (100 ml) and chloroform (100 ml) were added followed by cooling to a temperature of about 25 0C to about 30 0C. The pH of the reaction solution was adjusted to about 8.6 by the addition of acetic acid (1 1 ml). The organic and aqueous layers were separated and the aqueous layer was extracted with of chloroform (50 ml) and the organic layers were combined.To the combined organic layer was charged methyltertiarybutylether (450 ml) and stirred for about 45 minutes to about 60 minutes at a temperature of about 0 0C to about 5 0C. The separated solid was filtered and the solid was washed with methyltertiarybutylether (100 ml) to afford 46 g of a crude form of title compound.Sodium hydroxide flakes (10 g) and water (70 ml) were charged into a clean and dry round bottom flask and stirred for about 5 minutes to 10 minutes. The resultant solution was cooled to a temperature of about 200C to about 25 0C, followed by addition of the above-obtained wet solid. The resultant reaction mixture was stirred for about 25 minutes to about 30 minutes. Methanol (70 ml) was charged <n="22"/>to the reaction mixture followed by cooling to a temperature of about 10 0C to about 15 0C. The pH of the reaction solution was adjusted to about 9.3 by addition of acetic acid (14 ml). Water (230 ml) was added in to the reaction mixture and stirred for about 2 hours to 3 hours. The separated solid was filtered and the solid was washed with mixture solution of water (70 ml) and methanol (15 ml).The obtained wet solid was charged into a clean and dry round bottom flask containing water (230 ml) and stirred for about 5 minutes to about 10 minutes followed by addition of methanol (70 ml). The resultant reaction mixture was stirred for about 30 minutes to about 45 minutes. The solid was filtered and washed with methanol (15 ml) and water (70 ml) mixture solution and again with water (3 x 100 ml).Methyltertiarybutylether (270 ml) was charged into a clean and dry 4 neck round bottom flask followed by cooling to about 0 0C to about 5 0C. The obtained wet solid was dissolved in dichloromethane (80 ml) and the organic layer was separated. The obtained solution was added into a round bottom flask containing methyltertiarybutylether. The resultant reaction mixture was stirred for about 60 minutes to about 90 minutes at a temperature of about 0 0C to about 5 0C. The separated solid was filtered and the solid was washed with methyltertiarybutylether (15 ml) and suck dried for about 30 minutes to about 45 minutes.The obtained wet solid was slurry again in methyltertiarybutylether (170ml) for about 30 minutes to about 45 minutes at about 25 0C to about 30 0C. The solid filtered and washed with methyltertiarybutyl (170ml) ether and suck dried for about 45 minutes to about 60 minutes. The solid obtained was dried at about 45 0C to about 50 0C under vacuum for about 5 hours to about 6 hours to afford 29 g of the title compound. Purity By HPLC: 99.91 %. % of sulfone impurity: 0.047%. |
|
With 3-chloro-benzenecarboperoxoic acid; In chloroform; dimethyl sulfoxide; at -12.5 - -12℃; for 0.5h;Industry scale;Product distribution / selectivity; |
Chloroform (325 liters), and meta-chloro-per-benzoic-acid (39.0 kg) were taken into a reactor and the mixture was stirred for about 50 minutes. The meta- chloro-per-benzoic-acid layer which settles at the bottom was separated, and taken into an addition bulb. Chloroform (325 liters), 2-[[[4-(3-methoxypropoxy)-3-methyl-2- pyridinyl] methyl] thio]-H-benzimidazole (65 kg) and DMSO (130 liters) were taken into another reactor and cooled to a temperature of about -12.5 0C. The solution of meta-chloro-per-benzoic-acid prepared above was added to the cooled reaction mass slowly. The reaction mass was maintained at about -12 0C for about 30 minutes.A solution of water (325 liters) and sodium hydroxide (41 .6 kg) was added to the above reaction mass and stirred for about 10 minutes. The pH of the reaction mass was adjusted to about 8.5 to about 9.0 using acetic acid (44 liters). The organic layer was separated and the aqueous layer was extracted into chloroform (65 liters). The organic layer was then extracted into a solution of sodium hydroxide flakes (3.2 kg) in water (195 liters), followed by extraction with a solution of sodium hydroxide (2.0 kg) in water (130 liters). The combined aqueous layer was washed with chloroform (30 X 2 liters). The aqueous layer was given carbon treatment and filtered through a hyflow bed. The carbon bed was washed with water (65 liters). To the aqueous layer chloroform (65 liters) and methanol (65 liters) were added and the mixture cooled to about 22.5 0C. The pH of the reaction mixture was adjusted to about 8.5 to about 9.0 using a 1 :1 combination of acetic acid and water (20 liters), and the organic layer was separated, and the aqueous layer was extracted into chloroform (30 liters). The combined organic layer was added to methyl tertiary butyl ether (290 liters) cooled to a temperature of about 2 0C to about 5 0C. The reaction mass was maintained at about 2 0C to about 5 0C for about 15 minutes. The separated solid was filtered and washed with methyl tertiarybutyl ether (65 liters).The wet material and methanol (45 liters) were added to a solution of sodium hydroxide (6.5 kg) in water (45 liters) taken into a reactor. The reaction mass was stirred for about 25 minutes to about 30 minutes for clear dissolution and then cooled <n="24"/>to about 12.5 0C. The pH of the solution was adjusted to about 9.3 to about 9.7 using a 1 :1 solution of acetic acid in water (24 liters) followed by addition of water (98 liters). The pH was readjusted to about 9.3 to about 9.7 using a 1 :1 solution of acetic acid in water at about 12 0C to about 15 0C. The reaction mass was maintained at about 12 0C to about 15 0C for about 30 minutes. The separated solid was filtered and washed with a solution of water (45 liters) and methanol (10 liters). The wet solid was again slurried in a combination of water (215 liters) and methanol (45 liters) for about 45 minutes, and then filtered. The filtered solid was washed with a mixture of water (45 liters) and methanol (10 liters), followed by washing with water (195 liters). Metyl tertiary butyl ether (175 liters) was taken into a reactor and cooled to about 2.5 0C. Dichloromethane (52 liters) was added to it followed by addition of the wet material. The reaction mass was stirred for about 30 minutes at the same temperature and them filtered. The filtered material was washed with methyl tertiary butyl ether (10 liters).The wet material was taken into another 1 10 liters of methyl tertiary butyl ether and stirred for about 40 minutes. The material was then filtered and washed with methyl tertiary butyl ether (25 liters). The wet material was dried at about 47 0C for about 30 minutes to get 19.8 kg of the title compound. |
|
With trans-stilbene ozonide; In methanol; for 3.5h;Heating / reflux;Product distribution / selectivity; |
Trans-stilbene ozonide was prepared following the recipe of example 1, in an amount of 0.031 mol.The thus-prepared ozonide was added drop-wise over 0.5h to a boiling solution of sulfide (0.031 mol) in 100 ml of methanol. The reaction solution is boiled for 3 hours under reflux conditions. The experiment was performed for a number of sulfides, generally represented by formula III. After 3 hours, the reaction mixtures were analyzed for conversion rate. The results are shown in table 1. In all cases, sulfoxide selectivity was 100%. |
|
With 1-hexen-ozonide; In methanol; for 3.5h;Heating / reflux;Product distribution / selectivity; |
21 g 1-Hexene (0.20 mol) in 500 ml of methanol at -15C was converted with 1.2 molar equivalents of ozone, thus producing 1-hexen-ozonide. The ozonide was present as a clear solution in methanol.The solution was used directly for the oxidation of the sulfides represented by formula III. Thereto, the ozonide was added dropwise over 0.5h to the boiling solution of the sulfide (0.20 mol) in 200 ml methanol. The reaction solution was refluxed for 3 hours. After 3 hours, the reaction mixtures were analyzed for conversion rate. The results are shown in table 2. In all cases, sulfoxide selectivity was 100%. |
|
With dihydrogen peroxide;methyltrioxorhenium(VII); In 2,2,2-trifluoroethanol; at -20 - 0℃; |
Example 22: Synthesis and crystallization of rabeprazole sodium form F137.20 g (0.400 moles) Rabeprazole sulphide was dissolved in 360 ml 2, 2, 2-trifluoroethanol and cooled to about -200C. Then 34.3 ml of hydrogen peroxide 35% was added following with gradually addition of solution 200 mg methyltrioxorhenium (VII ) in 40 ml of 2 , 2 , 2-trifluoroethanol as catalyst for the reaction. The catalyst was added in portions. The mixture was stirred for up to 00C and the end of the reaction was determined upon in-process control compliance (HPLC assay for sulphide not more than 5 %) .After the reaction was completed 5 % aqueous solution of sodium thiosulphate were added at 10 -15C and pH of the solution was adjusted to 11 with 10 % aqueous solution of sodium hydroxide. 2, 2, 2-Trifluoroethanol was distilled off. 600 ml of acetonitrile/water mixture (1:3, v/v) was slowly added to the concentrated residue at about 15C and activated carbon was added to decolour the solution. The suspension was filtered through the randalite.Rabeprazole was isolated by precipitation with 50 % acetic acid. pH was adjusted to about 8.5 at about 15C to perform crystallization of rabeprazole, then cooled up to 00C. The crystallised product was collected by filtration, washed or macerated with acetonitrile . The product was dried in dryer below 45C, yield is 120 g.120.0 g of rabeprazole was suspended in 600 ml of acetonitrile at room temperature. Sodium salt was prepared with 26.8 g 50 % aqueous solution of sodium hydroxide and heating up to 500C to obtain a solution. Filtration was included to remove any un- dissolved particles. Rabeprazole sodium was precipitated with 600 ml diisopropyl ether which was slowly added to the solution at 20 +/- 3 0C. The crystallization was carried out for up to 10 hours at 20 +/- 3 0C, then gradually cooled up to 0 0C and left standing for about 5 hours. The crystallised product was collected by filtration and washed with diisopropyl ether. The product is dried in dryer below 45 0C. Yield is 122 g of rabeprazole sodium form F. |
|
With sodium hypochlorite; In dichloromethane; at 0 - 5℃; for 1.5h;Industry scale; |
2-[[[4-(3-Methoxypropoxy)~3-methyl-2-pyridinyl]methyl]thio]-1H- benzimidazole (130 Kg) was dissolved in methylene chloride(780 L)1 stirred at 25 - 350C and cooled the reaction mass to 0 - 50C. Sodium hypochlorite solution (2%, 975 L) was added to the reaction mass for 1 hour 30 minutes at 0 - 50C and stirred. Ammonium sulphate (122 Kg), water (1000 L), methylene chloride (250 L) and sodium chloride (130 Kg) were added to the reaction mass, stirred for 30 minutes at 5 - 100C and allowed to settle for 15 minutes. The bottom organic layer was separated twice by treating with methylene chloride (2 x 250 L), stirred and allowed to settle for 15 minutes. Water (1300 L) and sodium hydroxide flakes (50 Kg) were added to the reactor, cooled to 20 - 250C and then added the methylene chloride layer to the reactor. Sodium chloride (50 Kg) was added to the reaction mixture, stirred for 20 minutes and allowed to settle for 20 minutes. The bottom organic layer was separated. The pH of the aqueous layer was adjusted to 9.2 - 9.4 with ammonium acetate solution (ammonium acetate: 52 Kg + water: 200 L) and acetic acid solution (acetic acid: 65 L + water: 200 L). The bottom organic layer was separated twice by treating with methylene chloride (2 x 650 L), stirred and allowed to settle for 15 minutes. The organic layer was given carbon treatment, filtered and washed the filtrate with methylene chloride (50 L). Dried the total organic layers with sodium sulfate (20 Kg) and 2-Amino ethanol (1.8 L) was added. The organic layer was concentrated until the mass temperature reached to 40 - 450C. Acetonitrile (50 L) was added to the reaction mass and acetonitrile was distilled off from the reaction mass until the mass temperature reached to 40 - 450C. Acetonitrile (600 L) was added to the reaction mass, stirred for 2 hours at room temperature and cooled to 0 - 50C. Centrifuged the material twice, washed with acetonitrile (2 x 50 L) and dried to give 90 Kg of crude rabeprazole. Taken Monomethylamine (135 L) in water (765 L), cooled to 10 - 200C, added crude rabeprazole and stirred for dissolution. The bottom organic layer was separated twice by treating with methylene chloride (2 x 180 L), stirred and allowed to settle for 15 minutes. The pH of the aqueous layer was adjusted to 9.6 - 9.8 with ammonium acetate solution (ammonium acetate: 36 Kg + DM water: 140 L). The bottom organic layer was separated twice by treating with methylene chloride (2 x 450 L), stirred and allowed to settle for 15 minutes. The total organic layer was washed with sodiumchloride solution (sodium chloride: 36 Kg, DM water: 140 L), given carbon treatment, filtered, washed the filtrate with methylenechloride (50 L) and 2-amino ethanol (0.54 L) was added. The organic layer was concentrated until the mass temperature reached to 35 - 400C. Acetonitrile (90 L) was added to the reaction mass and acetonitrile was distilled off from the reaction mass until the mass temperature reached to 35 - 400C. Acetonitrile (270 L) was added to the reaction mass, cooled to 25 - 350C, stirred for 1 hour 30 minutes, cooled to 0 - 50C and stirred for 1 hour. Centrifuged the material, washed with acetonitrile (50 L) and dried the material to give 72 Kg of rabeprazole (HPLC purity: 99.8%). |
|
|
Example 1:Preparation of 2-{4-(3-Methoxypropoxy)-3-methylpyridine-2-yl} methy lsulfinyl-lH-benzimidazole (Rabeprazole) :To a RB flask IPA (30 mL) was charged followed by Rabeprazole sulfide (10 g) at 25-30 C. To this NaOH (2.0 equivalent) solution was added followed by the lot wise addition of 6%-12% NaOCl solution (>1.3 equivalent) at 0-5C. The reaction was monitored by HPLC. After completion of the reaction, reaction mass was poured into aqueous sodium thiosulphate pentahydrate solution (35-60%). The reaction solution subjected to carbon treatment and filtered. To the obtained filtrate, 2V MDC is charged, and then pH was adjusted to 8-8.8 using formic acid. Layers were separated and MDC distilled out under vacuum and stripped out with ethyl acetate. To the residue ethyl acetate was charged and cooled to 0-5 C. The solid obtained was filtered and washed with ethyl acetate. The crude Rabeprazole (Purity by HPLC: <99.0%) obtained was taken in ethyl acetate and treated with 1.0 equivalent of diethylamine at 50-55 C. To the clear solution Rabeprazole is seeded and the reaction mass cooled to 0-5 C. The solid obtained was filtered, washed with ethyl acetate to yield the pure Rabeprazole (Purity by HPLC: >99.7%) which was dried under vacuum at 50-55 C. |
|
With sodium hypochlorite; sodium hydroxide; In water; acetonitrile; at -8 - -2℃; |
Example 3 : Preparation of rabeprazole (Formula II) 2-( { [4-(3 -Methoxypropoxy)-3 -methylpyridin-2-yl]methyl } sulfanyl)- lH-benzimidazole (Formula VII) obtained in Example 2 (125 g) was added to acetonitrile (625 mL) and the reaction mixture was cooled to 10C to 15C. Sodium hydroxide solution (prepared by dissolving 29.2 g of sodium hydroxide in 475 mL of de-ionized water) was added to the reaction mixture at 8C to 15C for 5 minutes to 10 minutes. The reaction mixture was stirred at 8C to 15C and cooled to -7C to -8C. Sodium hypochlorite (1.05 mole equivalent) was added to the reaction mixture at -8C to -2C for 30 minutes to 50 minutes. Sodium thiosulphate solution (prepared by dissolving 12.5 g sodium thiosulphate in 125 mL de-ionized water) was added to the reaction mixture at -8C to +5C. The pH of the mixture was adjusted to 8.7 to 9.1 with aqueous acetic acid solution (80 mL; prepared by dissolving 40 mL glacial acetic acid with 40 mL de-ionized water) at 0C to 5C. Ethyl acetate (625 mL) was added to the mixture and the temperature was increased to 10C to 20C. The mixture was allowed to settle for 10 minutes to 30 minutes and the layers obtained were separated. The organic layer was concentrated at 15C to 30C and 20 mm to 30 mm of Hg pressure. The residue obtained was dissolved in acetone (625 mL) at 25C to 30C and the reaction mixture was cooled to 0C to 5C. Rabeprazole seed (0.25 g) was added to the reaction mixture and the reaction mixture was stirred at 0C to 5C. Diisopropyl ether (625 mL) was added to the mixture at 0C to 5C for 30 minutes to 45 minutes. The reaction mixture was stirred at 0C to 5C for 60 minutes to 70 minutes. The solid obtained was filtered and washed with pre-cooled (0C to 5C) acetone-diisopropyl ether (2 X 125 mL; prepared by dissolving 125 mL acetone in 125 mL diisopropyl ether). The solid obtained was dried at 40C to 45C at 10 mm to 20 mm of Hg pressure till the loss on drying was not more than 1.0% w/w to obtain the title compound. Yield: 0.72 w/w |
|
|
A solution of 10 - ((0.029mol) 2-[[[4-(3-methoxypropoxy)-3-methyl-2-pyridyl]-methyl]thio]-1H-benzimidazole (condensate) Add 100ml toluene, 0.2 g of purified water, 8. lg (+) - L-diethyl tartrate, 4.5 g of titanium tetraisopropoxide, 52-55 C for lh; cooled to 25 C, 3.4 g of N, N-diisopropylethylamine, 4.5 g hydrogen peroxide cumene, 25-30 C reaction 2 h. After the oxidation reaction, with 2.5% sodium hydroxide solution extraction 2 times, each 60ml, combined with sodium hydroxide solution, Add lg algae and lg activated carbon decolorization, the filtrate with saturated ammonium chloride solution to rhoEta = 9.7, the control temperature of 10-20 C, precipitation of white solid, crystallization lh after filtration. A 16 g dextroride dexeprazole was obtained (dry 8 g (0.022 mol 1)). 120 ml of dichloromethane and 300 ml of n-hexane were mixed and 0.88 g (0.022 mol) of sodium hydroxide solid was added with stirring. The control temperature is 10-20 C, Add dextrorolidazole wet to the above mixture, add finished, continue to control temperature 10-20 C 4h. The reaction was completed, filtered, washed, vacuum dried at 60 C, and dexamethasone was dissolved in sodium 7.2 g, yield: 72% Purity: 99.81%, sulfone:0.01%. |
|
|
20.59 g of 2 - [[4- (3-methoxypropoxy) -3-methylpyridin-2- yl] methyl] thio] -1H-benzimidazole and 11.81 g of tartaric acid derivative were placed in a solution of tris In the flask,Take 120 ml of toluene as a solvent.Heated to 120 , toluene with water for two hours.Then cooled to 90 C,5.33 ml of tetraisopropyl titanate was added while keeping warm,Stirred for 1.5 hours,Then add 0.324 ml of distilled water,Continue stirring insulation 1.5 hours,Then 3.0 ml of N, N-diisopropylethylamine was added,After stirring for 0.5 hour,The reaction system was cooled to 30 C.22.2 ml of phenylisopropylperoxide was added dropwise,After stirring at 30 C for 1.5 hours,Determination of enantiomeric excess and rabeprazole by HPLC method. 60 ml of a 30% aqueous sodium thiosulfate solution was added to the reaction solution,After stirring for 40 minutes, the toluene layer was separated,Toluene layer was added aqueous sodium hydroxide solution,Stirred for 1 hour,The toluene layer is extracted with water,Separate the water phase.The combined aqueous phases were washed twice with dichloromethane,Separate the water phase,The combined aqueous phase was added dropwise with sodium dihydrogen phosphate solution,Adjust the pH to precipitate a solid,Ice bath for 2 hours,filter,The filter cake was washed with distilled water,Sampling measurements and enantiomeric excess. The above solid was dissolved in acetone,Add water,Precipitation of the solid,Stir at room temperature for 2 hours.filter,Weigh 25.82 grams.Sampling measurements and enantiomeric excess,Its enantiomeric excess is 100% e.e,Sulfoxide content of 99.33%.The resulting solid was dissolved in acetone,And protected with nitrogen,Add water,Precipitation of the solid,After stirring at room temperature for 2 hours,Sampling content measurement,Its sulfoxideThe content is 99.77%. |
|
With sodium hydroxide; sodium hypochlorite; In methanol; water; at -25 - -15℃; for 0.783333 - 1.25h; |
To a solution of 2-[[4-(3-Methoxypropxy)-3-methylpyridine-2-yl]-2- methylthio]- l H-bezamidazole (Rabeprazole sulfide) (25 g) in methanol (125 ml), added sodium hydroxide solution (I.5N, 200 ml). Reaction mass was cooled to -15 to -25C under stirring. A solution of sodium hypochlorite (~1%, 550 ml) was added slowly at -15 to -25C to the reaction mass over a period of 45-75 minutes. The progress of reaction was monitored by HPLC. After completion of reaction, added solution of sodium thiosulphate and gradually raised the temperature to 1O0C in 20-30 minutes. The pH of reaction mass was adjusted to 12.8 to 13.5 using caustic solution (~ 50 ml) and mixture of methylene chloride (125 ml) & dimethyl acetamide (1.25ml) was added at room temperature and stirred for 15 minutes. Separated the two layers & kept methylene chloride layer aside. Again washed the aqueous layer with methylene chloride (75 ml) & dimethyl acetamide (1 ml) and separated the aqueous layer. Combined both methylene chloride layers and extracted with water (2 x 125 ml). The pH of combined aqueous layer was adjusted to 9.4 - 9.8 with saturated solution of ammonium chloride. Aqueous layer was extracted using methylene chloride (125ml) & dimethyl acetamide (ImI) followed by further extraction using a mixture of methylene chloride (75 ml) & dimethyl acetamide (1 ml). Combined both methylene chloride layers and washed with water (50 ml) .The methylene chloride layer was treated with activated carbon and dried (Na2SO4). To the residue obtained after distillation of methylene chloride completely under reduced pressure, added methanol (25 ml) and stirred for 5-10 minutes. The obtained solution was precipitated with diisopropyl ether (350 ml). The reaction mass was stirred for 1-2 hours at 20-300C and filtered .The wet cake was washed with diisopropyl ether (2 x 25 ml) and the product was dried under vacuum at 40-450CDry weight = 54 g. HPLC purity = 99.69% |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping