Abstract: In this study, we developed a simple transition-metal-free borylation reaction of aryl bromides. Bis-boronic acid (BBA), was used, and the borylation reaction was performed using a simple procedure at a mild temperature. Under mild conditions, aryl bromides were converted to arylboronic acids directly without any deprotection steps and purified by conversion to trifluoroborate salts. The functional group tolerance was considerably high. The mechanism study suggested that this borylation reaction proceeds via a radical pathway.
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
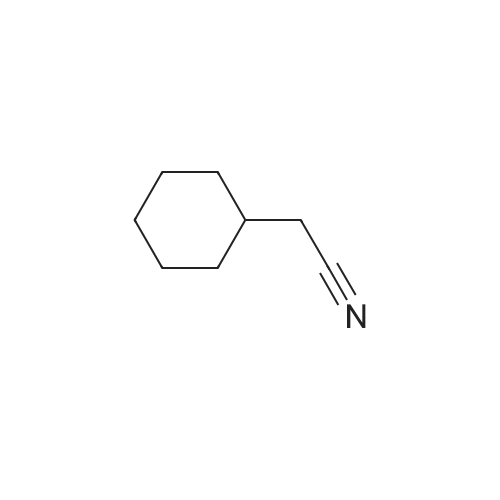
A. Cyclohexyl acetonitrile A solution of sodium cyanide (2.77 g, 56.5 mmol) in 70 ml of DMF was treated with cyclohexyl bromide (7.9 ml, 56.5 mmol), under N2. The reaction mixture was reacted for 48 hours and then partitioned between EtOAc and H2 O. The resultant layers were separated and the organic layer was concentrated in vacuo. The crude material was purified using flash chromatography (eluent of 5% EtOAc in hexanes). Yield: 5.0 g of a clear liquid (72%). 1 H NMR (d6 -DMSO): delta0.65-1.30 (m, 5H), 1.35-1.80 (m, 6H), 2.38 (d, 2H).
In ethyl acetate; N,N-dimethyl-formamide;
A. Cyclohexyl acetonitrile A solution of sodium cyanide (2.77 g, 56.5 mmol) in 70 ml of DMF was treated with cyclohexyl bromide (7.9 ml, 56.5 mmol), under N2. The reaction mixture was reacted for 48 hours and then partitioned between EtOAc and H2O. The resultant layers were separated and the organic layer was concentrated in vacuo. The crude material was purified using flash chromatography (eluent of 5% EtOAc in hexanes). Yield: 5.0 g of a clear liquid (72%). 1H NMR (d6-DMSO): delta 0.65-1.30 (m, 5H), 1.35-1.80 (m, 6H), 2.38 (d, 2H).
With hydrogenchloride; nitrogen; In ethanol; cyclohexane; benzene;
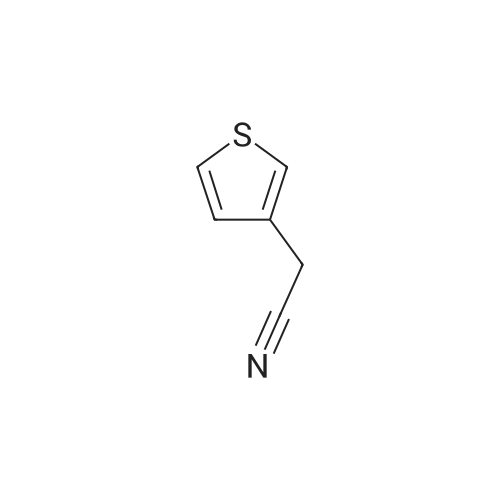
EXAMPLE 1 Preparation of α-(3-thienyl)-α-cyclohexyl-acetonitrile In a 100 ml-flask fitted with a mechanical stirrer, a vertical condensor protected by a calcium chloride stopper, a dropping-funnel and a source of nitrogen were introduced 30 ml of hexamethylenephosphotriamide and 2.3 g (0.1 mol) of finely cut sodium wire. A mixture of 12.3 g (0.1 mol) of (3-thienyl)-acetonitrile and 16.3 g (0.1 mol) of cyclohexyl bromide was then quickly added at a temperature of 20 C. The reaction mixture was then maintained under nitrogen atmosphere and stirred for 12 hours at room-temperature. The excess of sodium was destroyed by adding 5 ml of ethanol and the organic solution was slowly poured into 100 ml of a 1N iced solution of hydrochloric acid. The solution was extracted twice with 100 ml ether. The ethereal phases were collected, washed with water, dried and concentrated under reduced pressure. The crude product was then purified by chromatography on a silica column (150 g of silica) using a 1/1 benzene/cyclohexane mixture as elution agent. The product obtained was rectified by distillation. In this manner, 3.4 g of α-(3-thienyl)-α-cyclohexyl-acetonitrile were obtained, which represents a yield of 16%. B.P. 130 C under 3 mm Hg.
With hydrogenchloride; In ethanol; cyclohexane; benzene;
EXAMPLE 2 Preparation of α-(3-thienyl)-α-cyclohexyl-acetonitrile In a flask fitted as in Example 1 hereabove, were placed 350 ml of anhydrous benzene, 86 g (0.7 mol) of (3-thienyl)-acetonitrile and 114 g (0.7 mol) of cyclohexyl bromide. Under nitrogen atmosphere, a suspension of 27 g of sodium amide in 250 ml of anhydrous benzene were added to the mixture. The reaction was exothermic and, for this reason, the sodium amide was introduced into the reaction medium slowly so that the temperature was maintained between 20 and 25 C. After this operation, the reaction medium was stirred for three hours at room-temperature. Then, 50 ml of ethanol were added and the solution was slowly poured into 1 liter of a 1N solution of hydrochloric acid. The benzene phase was decanted and the aqueous phase was extracted with 500 ml of ether. The organic phases were collected, washed with water, dried and concentrated under reduced pressure. The crude product was then purified by chromatography on a column of silica (800 g of silica) using a 1/1 benzene/cyclohexane mixture as elution agent. In this manner, 75 g of α-(3-thienyl)-α-cyclohexyl-acetonitrile were obtained, which represents a yield of 52%.
With hydrogenchloride; In N-methyl-acetamide;
EXAMPLE 3 Preparation of α-(3-thienyl)-α-cyclohexyl-acetonitrile In a 100 ml-flask fitted as in Example 1 hereabove, were placed 45 ml of dimethylformamide and 4 g (0.165 mol) of a suspension of sodium hydride in oil. The reaction mixture was cooled to -20 C by means of a bath comprising acetone and carbon dioxide ice and, then a mixture of 27 g (0.165 mol) of cyclohexyl bromide and 20 g (0.15 mol) of (3-thienyl)-acetonitrile was slowly added under nitrogen atmosphere. The reaction was strongly exothermic and the medium was maintained at a temperature of -20 C. Stirring was continued for two hours at -20 C, after which the temperature was allowed to return slowly to room-temperature, the mixture being stirred and maintained under nitrogen atmosphere all the time. The reaction was allowed to continue for 12 hours at +20 C and then the mixture was poured into 150 ml of a 1N iced solution of hydrochloric acid. The aqueous solution was extracted twice with 100 ml of ether, the ethereal phases were collected, washed with water, dried and concentrated under reduced pressure. The crude product so obtained was then distilled. In this manner, 16 g of α-(3-thienyl)-α-cyclohexyl-acetonitrile were obtained, which represents a yield of 50%. B.P. 130-135 C under 3 mm Hg. Following the same procedure as that described above but using the appropriate starting-products the compound hereunder was prepared:
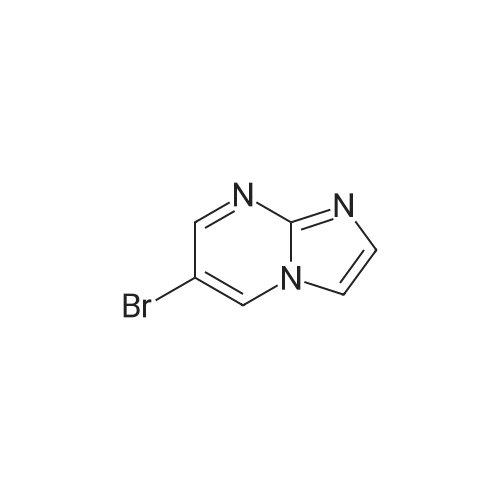
12 ml of ethanol, 920 mg of potassium hydroxide pellets and 1 g of <strong>[865156-68-9]6-bromoimidazo[1,2-a]pyrimidine</strong> are charged to a glass tube. The tube is sealed and heated at 135 C. for 12 minutes using microwave radiation. After returning to a temperature in the vicinity of 20 C., 1.5 ml of bromocyclohexane are added. The tube is again sealed and the combined mixture is heated at 140 C. for 15 minutes using microwave radiation. After returning to a temperature in the vicinity of 20 C., the reaction medium is evaporated to dryness under reduced pressure and the solid isolated is chromatographed, under argon pressure, on silica gel (eluent dichloromethane/methanol 97/3). 75 mg of 6-(cyclohexyloxy)imidazo[1,2-a]pyrimidine are thus obtained in the form of a beige solid. MS: method B; [M+H]+ m/z=218; Tr=2.54 min.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping