| 56% |
|
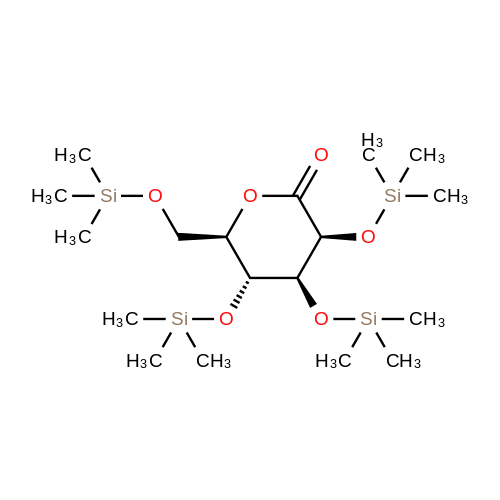
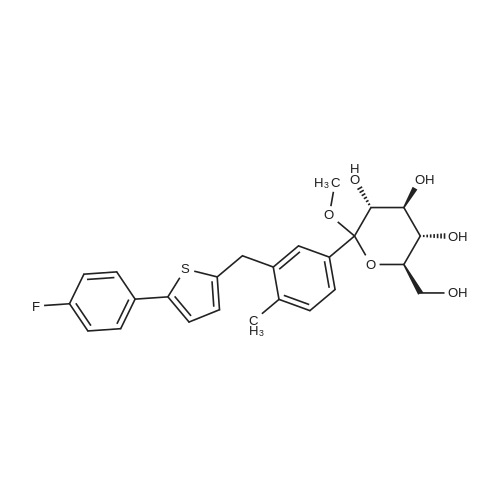
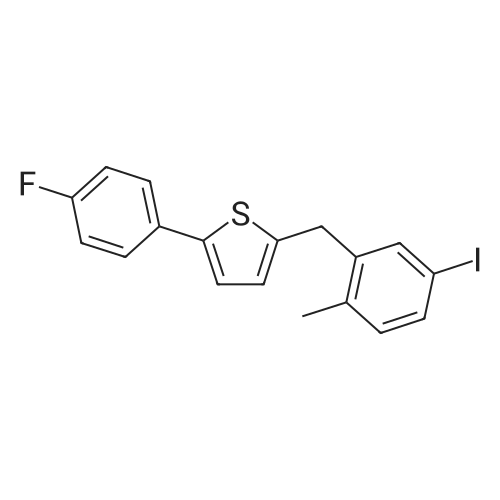
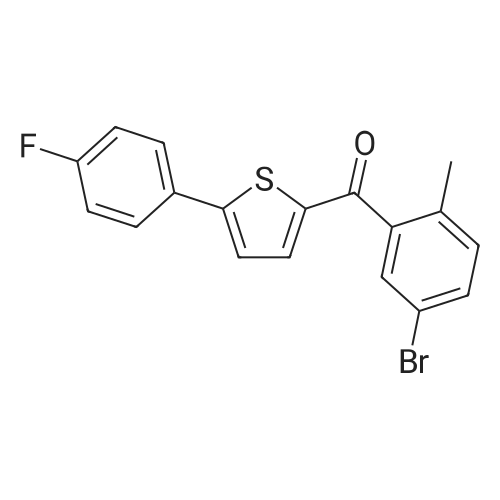
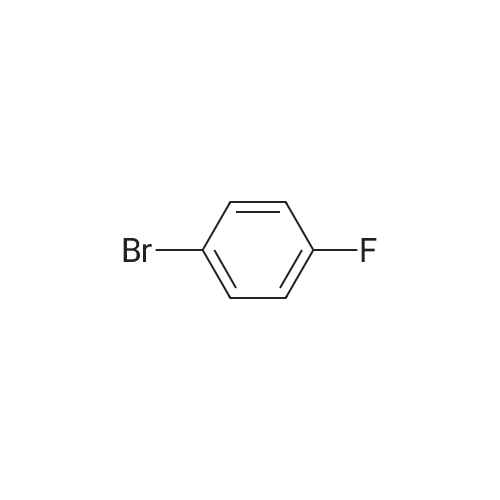
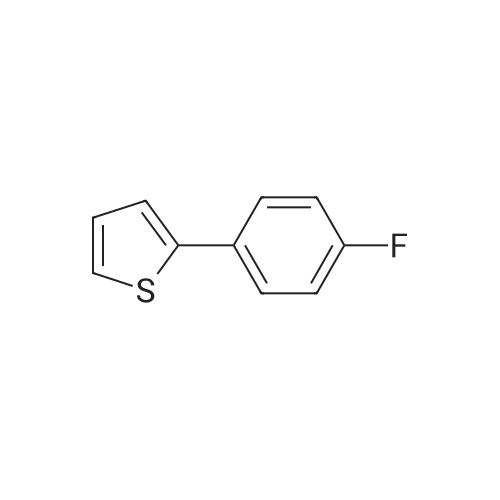
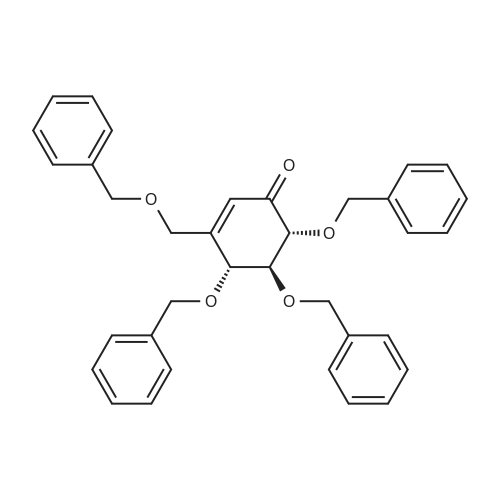
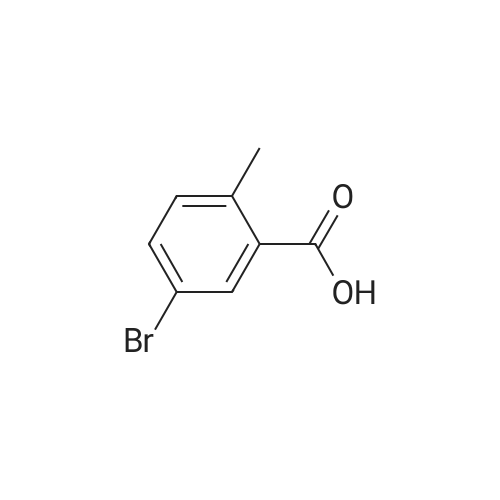
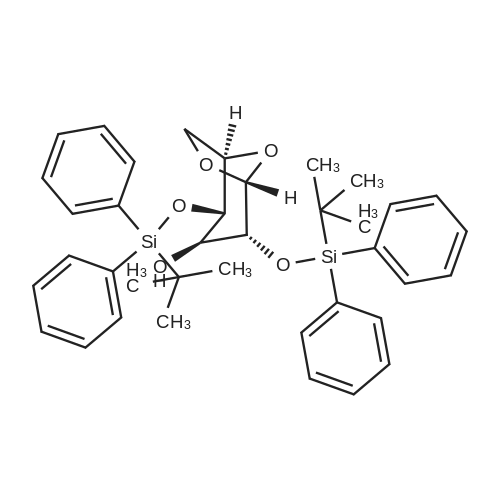
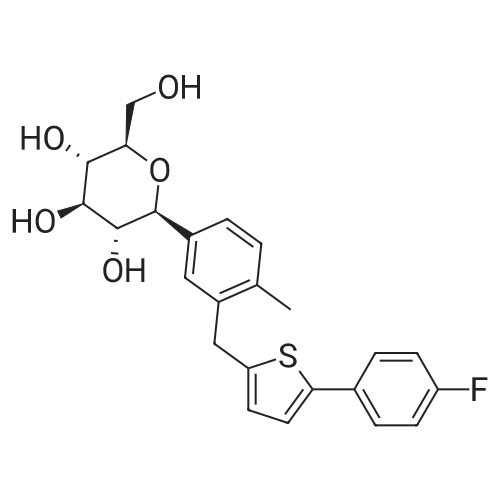
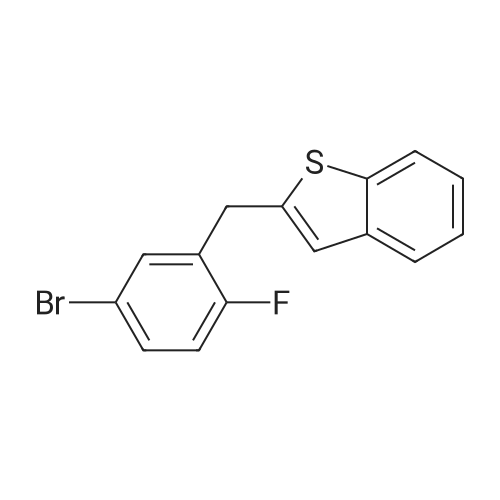
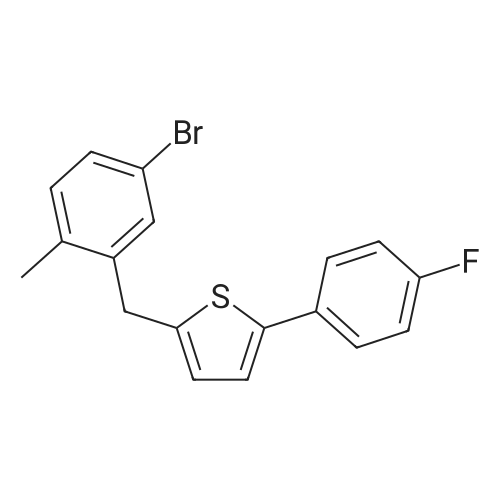
2-(5-Bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (1.5 g, 4.15 mmol) and magnesium powder (0.33 g, 13.7 mmol) were placed in a suitable reactor, followed by THF (9 mL) and 1,2-dibromoethane (95 muL). The mixture was heated to reflux. After the reaction was initiated, a solution of 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (2.5 g, 6.92 mmol) in THF (15 mL) was added dropwise. The mixture was stirred for another 2 hours under reflux, and was then cooled to ambient temperature and titrated to determine the concentration. The thus prepared 3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl magnesium bromide (0.29 M in THF, 17 mL, 5.0 mmol) and AlCl3 (0.5 M in THF, 4.0 mL, 2.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of 1,6-anhydro-2,4-di-O-tert-butyldiphenylsilyl-beta-D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added n-BuLi (0.4 mL, 1.0 mmol, 2.5 M solution in Bu2O). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60° C. (external bath temperature) to remove low-boiling point ethereal solvents, and PhOMe (6 mL) was then added. The remaining mixture was heated at 150° C. (external bath temperature) for 5 hours at which time HPLC assay analysis indicated a 68percent yield of 2,4-di-O-tert-butyldiphenylsilyl-1-C-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-beta-D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10percent aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:20 MTBE/n-heptane) to give the product 2,4-di-O-tert-butyldiphenylsilyl-1-C-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-beta-D-glucopyranoside (0.51 g, 56percent) as a white powder. [0273] 1H NMR (400 MHz, CDCl3) delta 7.65 (d, J=7.2 Hz, 2H), 7.55 (d, J=7.2 Hz, 2H), 7.48 (dd, J=7.6, 5.6 Hz, 2H), 7.44-7.20 (m, 16H), 7.11-6.95 (m, 6H), 6.57 (d, J=3.2 Hz, 1H), 4.25 (d, J=9.6 Hz, 1H), 4.06 (s, 2H), 3.90-3.86 (m, 1H), 3.81-3.76 (m, 1H), 3.61-3.57 (m, 1H), 3.54-3.49 (m, 2H), 3.40 (dd, J=8.8, 8.8 Hz, 1H), 2.31 (s, 3H), 1.81 (dd, J=6.6, 6.6 Hz, 1H, OH), 1.19 (d, J=4.4 Hz, 1H, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDCl3) delta 162.1 (d, J=246 Hz, C), 143.1 (C), 141.4 (C), 137.9 (C), 136.8 (C), 136.5 (C), 136.4 (CH×2), 136.1 (CH×2), 135.25 (C), 135.20 (CH×2), 135.0 (CH×2), 134.8 (C), 132.8 (C), 132.3 (C), 130.9 (d, J=3.5 Hz, C), 130.5 (CH), 130.0 (CH), 129.7 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.6 (CH×4), 127.5 (CH×2), 127.2 (CH×2), 127.1 (d, J=8.2 Hz, CH×2), 127.06 (CH), 126.0 (CH), 122.7 (CH), 115.7 (d, J=21.8 Hz, CH×2), 82.7 (CH), 80.5 (CH), 79.4 (CH), 76.3 (CH), 72.9 (CH), 62.8 (CH2), 34.1 (CH2), 27.2 (CH3×3), 26.7 (CH3×3), 19.6, (C), 19.3 (CH3), 19.2 (C); LCMS (ESI) m/z 938 (100, [M+NH4]+), 943 (10, [M+Na]+). 2 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping